

Structure, mechanism and evolution of enzymes
Protein archaeology to synthetic biology
We use the power of evolution to create new enzymes, and combine these enzymes to make new metabolic systems. From the earliest proteins to modern synthetic biology and chemical biology, understanding evolution at the molecular level is fundamental to engineering biology.
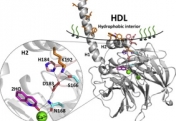
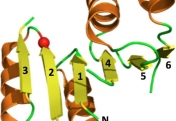
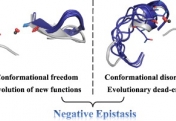
How do proteins evolve? Natural selection can create molecular machines with breath-taking performances, e.g., enzymes that accelerate the rate of chemical transformations by factors of 106 up to 1017. Strikingly, new protein functions can evolve within years or even months, as happens with drug resistance, and with enzymes that degrade man-made chemicals. Why is this process, which is based on ‘trial and error’ so rapid and efficient? We lack the fossils of the protein world, but we can reproduce protein evolution in the laboratory and in real time, implementing the principles of Darwinian evolution to individual genes and enzymes. In doing so, we obtain crucial insights regarding the evolutionary intermediates and the routes and mechanisms that led to the highly proficient enzymes known to us today.
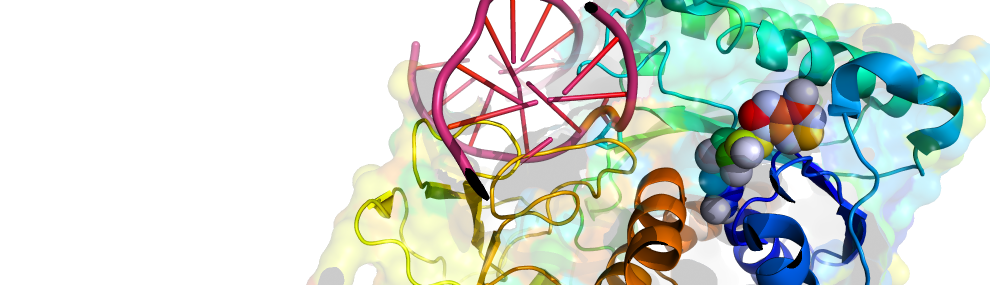
Our scientific approach is interdisciplinary, combining bioinformatics and molecular evolution, molecular and structural biology, biochemistry and organic chemistry. The enzymes we study originate from bacteria, human, plants and marine organisms. Their functions vary - hydrolases, dehydrogenases, methyltransferases, lyases and isomerases. Their time of emergence ranges from several decades (enzymes that evolved to degrade man-made chemicals and drugs) to ~3.7 billion years ago (the first protein enzymes).
What we understand, we can make: Knowledge derived from the reconstruction of past evolutionary events enables us engineer new enzymes with tailor-made properties, for various applications including. nerve agent detoxification or improved crop yields.