

Publications
2025
-
(2025) Nature Cell Biology. 27, p. 1887-1888 Abstract
Phase separation is a mechanism for non-organellar macromolecule segregation typical in the cell cytosol and nucleus. Two recent studies revealed functional phase separation within the endoplasmic reticulum, where calcium-mediated condensates co-ordinate chaperones and disulfide catalysts to enhance secretory protein production.
-
(2025) Proteins: Structure, Function and Bioinformatics. 94, 1, p. 25-50 Abstract[All authors]
This article presents an in-depth analysis of selected CASP16 targets, with a focus on their biological and functional significance. The authors highlight the most relevant features of the target proteins and discuss how well these were reproduced in the submitted predictions. While the overall performance of structure prediction methods remains impressive, challenges persist, particularly in modeling rare structural motifs, flexible regions, small molecule interactions, posttranslational modifications, and biologically important interfaces. Addressing these limitations can strengthen the role of structure prediction in complementing experimental efforts and advancing both basic research and biomedical applications.
-
(2025) Inorganic Chemistry. 64, 11, p. 5568-5578 Abstract
Mucin glycoproteins are secreted from epithelial goblet cells to create protective barriers lining the intestines, stomach, lungs, and other body surfaces. MUC2 is the primary glycoprotein secreted in the intestine and is essential for intestinal homeostasis. The D1 segment of the MUC2 N-terminal region was recently shown to bind Cu2+ and Cu+ separately in a unique two-tiered binding site. Copper is an essential metal acquired through diet for cells and enzymes to function properly, but little is known about how it is handled in the digestive tract. With both oxidation states of Cu in the intestine, we asked how the binding of Cu+ to MUC2 D1 impacts the binding of Cu2+ and vice versa. Here, we use a combination of competition titrations, electron paramagnetic spectroscopy, and X-ray absorption spectroscopy to characterize the physical properties of Cu2+ and Cu+ binding to MUC2 D1 at pH values relevant to the intestine. Our data show that simultaneous yet noncooperative binding of Cu2+ and Cu+ is possible and further reveal new insights into the pH dependence and plasticity of the Cu2+ and Cu+ binding sites. These results inspire interesting questions about the functional roles of MUC2 Cu handling in the intestinal tract.
-
(2025) Proceedings of the National Academy of Sciences - PNAS. 122, 10, e241971712. Abstract
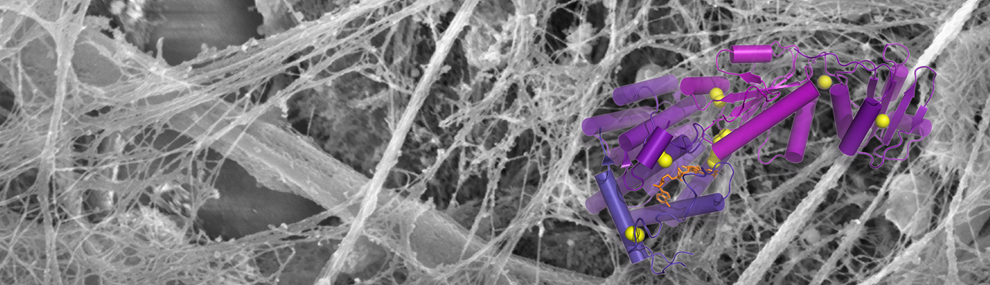
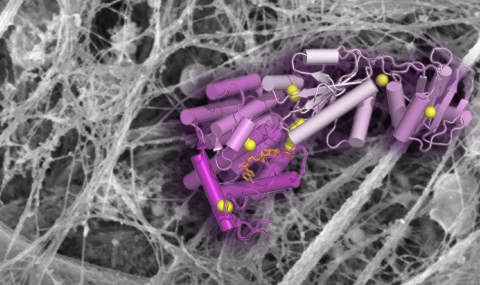
Secreted mucins are multimegadalton glycoprotein polymers that share the function of protecting mucosal tissues but diversified for activities in different organs of the body. Structural studies of secreted mucins are complicated by the enormous sizes, flexibility, and complex supramolecular assembly modes of these glycoproteins. The two major respiratory mucins are MUC5AC and MUC5B. Here, we present structures of a large amino-terminal segment of MUC5AC in the form of helical filaments. These filaments differ from filamentous and tubular structures observed previously for the intestinal mucin MUC2 and the partial mucin homolog VWF. Nevertheless, the MUC5AC helical filaments support the proposed mechanism, based on MUC2 and VWF, for how noncovalent interactions between mucin monomers guide disulfide crosslinking to form polymers. The high-resolution MUC5AC structures show how local and limited changes in amino acid sequence can profoundly affect higher-order assembly while preserving the overall folds and polymerization activity of mucin glycoproteins. Differences in supramolecular assembly are likely to be functionally significant considering the divergence of mechanical properties and physiological requirements between respiratory and intestinal mucins. Determining the high-resolution structures of respiratory mucins provides a foundation for understanding the mechanisms by which they clean and protect the lungs. Moreover, the MUC5AC structure enables visualization of the sites of human amino acid sequence variation and disease-associated mutations.
2024
-
(2024) Protein Science. 33, 3, e4929. Abstract
Domains known as von Willebrand factor type D (VWD) are found in extracellular and cell-surface proteins including von Willebrand factor, mucins, and various signaling molecules and receptors. Many VWD domains have a glycine-aspartate-proline-histidine (GDPH) amino-acid sequence motif, which is hydrolytically cleaved post-translationally between the aspartate (Asp) and proline (Pro). The Fc IgG binding protein (FCGBP), found in intestinal mucus secretions and other extracellular environments, contains 13 VWD domains, 11 of which have a GDPH cleavage site. In this study, we investigated the structural and biophysical consequences of Asp-Pro peptide cleavage in a representative FCGBP VWD domain. We found that endogenous Asp-Pro cleavage increases the resistance of the domain to exogenous proteolytic degradation. Tertiary structural interactions made by the newly generated chain termini, as revealed by a crystal structure of an FCGBP segment containing the VWD domain, may explain this observation. Notably, the Gly-Asp peptide bond, upstream of the cleavage site, assumed the cis configuration in the structure. In addition to these local features of the cleavage site, a global organizational difference was seen when comparing the FCGBP segment structure with the numerous other structures containing the same set of domains. Together, these data illuminate the outcome of GDPH cleavage and demonstrate the plasticity of proteins with VWD domains, which may contribute to their evolution for function in a dynamic extracellular environment.
-
(2024) Nature Biomedical Engineering. 8, 1, p. 30-44 Abstract[All authors]
Conventional methods for humanizing animal-derived antibodies involve grafting their complementarity-determining regions onto homologous human framework regions. However, this process can substantially lower antibody stability and antigen-binding affinity, and requires iterative mutational fine-tuning to recover the original antibody properties. Here we report a computational method for the systematic grafting of animal complementarity-determining regions onto thousands of human frameworks. The method, which we named CUMAb (for computational human antibody design; available at http://CUMAb.weizmann.ac.il), starts from an experimental or model antibody structure and uses Rosetta atomistic simulations to select designs by energy and structural integrity. CUMAb-designed humanized versions of five antibodies exhibited similar affinities to those of the parental animal antibodies, with some designs showing marked improvement in stability. We also show that (1) non-homologous frameworks are often preferred to highest-homology frameworks, and (2) several CUMAb designs that differ by dozens of mutations and that use different human frameworks are functionally equivalent.
2023
-
(2023) FEBS Journal. 290, 21, p. 5196-5203 Abstract
CysD domains are disulfide-rich modules embedded within long O-glycosylated regions of mucin glycoproteins. CysD domains are thought to mediate intermolecular adhesion during the intracellular bioassembly of mucin polymers and perhaps also after secretion in extracellular mucus hydrogels. The human genome encodes 18 CysD domains distributed across three different mucins. To date, experimental structural information is available only for the first CysD domain (CysD1) of the intestinal mucin MUC2, which is one of the most divergent of the CysDs. To provide experimental data on a CysD that is representative of a larger branch of the fold family, we determined the crystal structure of the seventh CysD domain (CysD7) from MUC5AC, a mucin found in the respiratory tract and stomach. The MUC5AC CysD7 structure revealed a single calcium-binding site, contrasting with the two sites in MUC2 CysD1. The MUC5AC CysD7 structure also contained an additional α-helix absent from MUC2 CysD1, with potential functional implications for intermolecular interactions. Lastly, the experimental structure emphasized the flexibility of the loop analogous to the main adhesion loop of MUC2 CysD1, suggesting that both sequence divergence and physical plasticity in this region may contribute to the adaptation of mucin CysD domains.
-
(2023) Trends in Pharmacological Sciences. 44, 11, p. 755-757 Abstract
Aberrant expression of transmem-brane mucins promotes tumor progression and interferes with im-munological and medicinal elimina-tion of cancer cells. In a recent article, Pedram et al. directed an at-tenuated bacterial mucin-specific protease to HER2-positive tumor cells and observed decreased tumor growth rates and extended survival of mice bearing HER2-positive tumors.
-
-
(2023) Current Opinion in Structural Biology. 79, 102524. Abstract
Contrary to first appearances, mucus structural biology is not an oxymoron. Though mucus hydrogels derive their characteristics largely from intrinsically disordered, heavily glycosylated polypeptide segments, the secreted mucin glycoproteins that constitute mucus undergo an orderly assembly process controlled by folded domains at their termini. Recent structural studies revealed how mucin complexes promote disulphide-mediated polymerization to produce the mucus gel scaffold. Additional protein-protein and protein-glycan interactions likely tune the mesoscale properties, stability, and activities of mucins. Evidence is emerging that even intrinsically disordered glycosylated segments have specific structural roles in the production and properties of mucus. Though soft-matter biophysical approaches to understanding mucus remain highly relevant, high-resolution structural studies of mucins and other mucus components are providing new perspectives on these vital, protective hydrogels.
-
(2023) EMBO Journal. 42, 2, e111869. Abstract[All authors]
Mucus is made of enormous mucin glycoproteins that polymerize by disulfide crosslinking in the Golgi apparatus. QSOX1 is a catalyst of disulfide bond formation localized to the Golgi. Both QSOX1 and mucins are highly expressed in goblet cells of mucosal tissues, leading to the hypothesis that QSOX1 catalyzes disulfide-mediated mucin polymerization. We found that knockout mice lacking QSOX1 had impaired mucus barrier function due to production of defective mucus. However, an investigation on the molecular level revealed normal disulfide-mediated polymerization of mucins and related glycoproteins. Instead, we detected a drastic decrease in sialic acid in the gut mucus glycome of the QSOX1 knockout mice, leading to the discovery that QSOX1 forms regulatory disulfides in Golgi glycosyltransferases. Sialylation defects in the colon are known to cause colitis in humans. Here we show that QSOX1 redox control of sialylation is essential for maintaining mucosal function.
2022
-
(2022) Blood. 140, 26, p. 2835-2843 Abstract
The von Willebrand factor (VWF) glycoprotein is stored in tubular form in Weibel-Palade bodies (WPBs) before secretion from endothelial cells into the bloodstream. The organization of VWF in the tubules promotes formation of covalently linked VWF polymers and enables orderly secretion without polymer tangling. Recent studies have described the high-resolution structure of helical tubular cores formed in vitro by the D1D2 and DD3 amino-terminal protein segments of VWF. Here we show that formation of tubules with the helical geometry observed for VWF in intracellular WPBs requires also the VWA1 (A1) domain. We reconstituted VWF tubules from segments containing the A1 domain and discovered it to be inserted between helical turns of the tubule, altering helical parameters and explaining the increased robustness of tubule formation when A1 is present. The conclusion from this observation is that the A1 domain has a direct role in VWF assembly, along with its known activity in hemostasis after secretion.
-
(2022) FEBS Letters. 596, 22, p. 2859-2872 Abstract
Formation of disulfide bonds in secreted and cell-surface proteins involves numerous enzymes and chaperones abundant in the endoplasmic reticulum (ER), the first and main site for disulfide bonding in the secretory pathway. Although the Golgi apparatus is the major station after the ER, little is known about thiol-based redox activity in this compartment. QSOX1 and its paralog QSOX2 are the only known Golgi-resident enzymes catalyzing disulfide bonding. The localization of disulfide catalysts in an organelle downstream of the ER in the secretory pathway has long been puzzling. Recently, it has emerged that QSOX1 regulates particular glycosyltransferases, thereby influencing a central activity of the Golgi. Surprisingly, a few important disulfide-mediated multimerization events occurring in the Golgi were found to be independent of QSOX1. These multimerization events depend, however, on the low pH of the Golgi lumen and secretory granules. We compare and contrast disulfide-mediated multimerization in the ER vs. the Golgi to illustrate the variety of mechanisms controlling covalent supramolecular assembly of secreted proteins.
-
(2022) Cell. 185, 22, p. 4206-4215.e11 Abstract[All authors]
Mucus protects the epithelial cells of the digestive and respiratory tracts from pathogens and other hazards. Progress in determining the molecular mechanisms of mucus barrier function has been limited by the lack of high-resolution structural information on mucins, the giant, secreted, gel-forming glycoproteins that are the major constituents of mucus. Here, we report how mucin structures we determined enabled the discovery of an unanticipated protective role of mucus: managing the toxic transition metal copper. Using two juxtaposed copper binding sites, one for Cu2+ and the other for Cu1+, the intestinal mucin, MUC2, prevents copper toxicity by blocking futile redox cycling and the squandering of dietary antioxidants, while nevertheless permitting uptake of this important trace metal into cells. These findings emphasize the value of molecular structure in advancing mucosal biology, while introducing mucins, produced in massive quantities to guard extensive mucosal surfaces, as extracellular copper chaperones.
-
(2022) Proceedings of the National Academy of Sciences of the United States of America. 119, 15, e211679011. Abstract
The glycoprotein von Willebrand factor (VWF) contributes to hemostasis by stanching injuries in blood vessel walls. A distinctive feature of VWF is its assembly into long, helical tubules in endothelial cells prior to secretion. When VWF is released into the bloodstream, these tubules unfurl to release linear polymers that bind subendothelial collagen at wound sites, recruit platelets, and initiate the clotting cascade. VWF evolved from gel-forming mucins, the polymeric glycoproteins that coat and protect exposed epithelia. Despite the divergent function of VWF in blood vessel repair, sequence conservation and shared domain organization imply that VWF retained key aspects of the mucin bioassembly mechanism. Here, we show using cryo-electron microscopy that the ability to form tubules, a property hitherto thought to have arisen as a VWF adaptation to the vasculature, is a feature of the amino-terminal region of mucin. This segment of the human intestinal gel-forming mucin (MUC2) was found to self-assemble into tubules with a striking resemblance to those of VWF itself. To facilitate a comparison, we determined the residue-resolution structure of tubules formed by the homologous segment of VWF. The structures of the MUC2 and VWF tubules revealed the flexible joints and the intermolecular interactions required for tubule formation. Steric constraints in full-length MUC2 suggest that linear filaments, a previously observed supramolecular assembly form, are more likely than tubules to be the physiological mucin storage intermediate. Nevertheless, MUC2 tubules indicate a possible evolutionary origin for VWF tubules and elucidate design principles present in mucins and VWF.
2021
-
(2021) mBio. 12, 6, e02602-21. Abstract[All authors]
In the parasite Trypanosoma brucei, the causative agent of human African sleeping sickness, all mRNAs are trans-spliced to generate a common 59 exon derived from the spliced leader (SL) RNA. Perturbations of protein translocation across the endoplasmic reticulum (ER) induce the spliced leader RNA silencing (SLS) pathway. SLS activation is mediated by a serine-threonine kinase, PK3, which translocates from the cytosolic face of the ER to the nucleus, where it phosphorylates the TATA-binding protein TRF4, leading to the shutoff of SL RNA transcription, followed by induction of programmed cell death. Here, we demonstrate that SLS is also induced by depletion of the essential ER-resident chaperones BiP and calreticulin, ER oxidoreductin 1 (ERO1), and the Golgi complex-localized quiescin sulfhydryl oxidase (QSOX). Most strikingly, silencing of Rhomboid-like 1 (TIMRHOM1), involved in mitochondrial protein import, also induces SLS. The PK3 kinase, which integrates SLS signals, is modified by phosphorylation on multiple sites. To determine which of the phosphorylation events activate PK3, several individual mutations or their combination were generated. These mutations failed to completely eliminate the phosphorylation or translocation of the kinase to the nucleus. The structures of PK3 kinase and its ATP binding domain were therefore modeled. A conserved phenylalanine at position 771 was proposed to interact with ATP, and the PK3F771L mutation completely eliminated phosphorylation under SLS, suggesting that the activation involves most if not all of the phosphorylation sites. The study suggests that the SLS occurs broadly in response to failures in protein sorting, folding, or modification across multiple compartments.
-
(2021) FEBS Journal. 288, 22, p. 6465-6475 Abstract
Zymogen granule membrane protein 16 (ZG16) is produced in organs that secrete large quantities of enzymes and other proteins into the digestive tract. ZG16 binds microbial pathogens, and lower ZG16 expression levels correlate with colorectal cancer, but the physiological function of the protein is poorly understood. One prominent attribute of ZG16 is its ability to bind glycans, but other aspects of the protein may also contribute to activity. An intriguing feature of ZG16 is a CXXC motif at the carboxy terminus. Here, we describe crystal structures and biochemical studies showing that the CXXC motif is on a flexible tail, where it contributes little to structure or stability but is available to engage in redox reactions. Specifically, we demonstrate that the ZG16 cysteine thiols can be oxidized to a disulfide by quiescin sulfhydryl oxidase 1, which is a sulfhydryl oxidase present together with ZG16 in the Golgi apparatus and in mucus, as well as by protein disulfide isomerase. ZG16 crystal structures also draw attention to a nonproline cis peptide bond that can isomerize within the protein and to the mobility of glycine-rich loops in the glycan-binding site. An understanding of the properties of the ZG16 CXXC motif and the discovery of internal conformational switches extend existing knowledge relating to the glycan-binding activity of the protein.
2020
-
(2020) Proceedings of the National Academy of Sciences of the United States of America. 117, 44, p. 27374-27380 Abstract
The complex environment of biological cells and tissues has motivated development of three-dimensional (3D) imaging in both light and electron microscopies. To this end, one of the primary tools in fluorescence microscopy is that of computational deconvolution. Wide-field fluorescence images are often corrupted by haze due to out-of-focus light, i.e., to cross-talk between different object planes as represented in the 3D image. Using prior understanding of the image formation mechanism, it is possible to suppress the cross-talk and reassign the unfocused light to its proper source post facto. Electron tomography based on tilted projections also exhibits a cross-talk between distant planes due to the discrete angular sampling and limited tilt range. By use of a suitably synthesized 3D point spread function, we show here that deconvolution leads to similar improvements in volume data reconstructed from cryoscanning transmission electron tomography (CSTET), namely a dramatic in-plane noise reduction and improved representation of features in the axial dimension. Contrast enhancement is demonstrated first with colloidal gold particles and then in representative cryotomograms of intact cells. Deconvolution of CSTET data collected from the periphery of an intact nucleus revealed partially condensed, extended structures in interphase chromatin.
-
(2020) PLoS Computational Biology. 16, 10, e1008382. Abstract[All authors]
The funding statement for this article should read as follows: \u201cThe research was supported by grants from the European Research Council (335439 to SJF, 636752 to MS, and 310649 and 825076 to DF), the Israel Science Foundation to MS (300/ 17) and through its Center of Excellence in Structural Cell Biology to SJF and DF (1775/12), a research grant from Sheri and David E. Stone and by a charitable donation from Sam Switzer and family. M.S. is an incumbent of the Aharon and Ephraim Katzir Memorial Professorial Chair. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.\u201d
-
(2020) Antioxidants & Redox Signaling. 33, 10, p. 665-678 Abstract
Aims:The post-translational oxidation of methionine to methionine sulfoxide (MetSO) is a reversible process, enabling the repair of oxidative damage to proteins and the use of sulfoxidation as a regulatory switch. MetSO reductases catalyze the stereospecific reduction of MetSO. One of the mammalian MetSO reductases, MsrB3, has a signal sequence for entry into the endoplasmic reticulum (ER). In the ER, MsrB3 is expected to encounter a distinct redox environment compared with its paralogs in the cytosol, nucleus, and mitochondria. We sought to determine the location and arrangement of MsrB3 redox-active cysteines, which may couple MsrB3 activity to other redox events in the ER. Results:We determined the human MsrB3 structure by using X-ray crystallography. The structure revealed that a disulfide bond near the protein amino terminus is distant in space from the active site. Nevertheless, biochemical assays showed that these amino-terminal cysteines are oxidized by the MsrB3 active site after its reaction with MetSO. Innovation:This study reveals a mechanism to shuttle oxidizing equivalents from the primary MsrB3 active site toward the enzyme surface, where they would be available for further dithiol-disulfide exchange reactions. Conclusion:Conformational changes must occur during the MsrB3 catalytic cycle to transfer oxidizing equivalents from the active site to the amino-terminal redox-active disulfide. The accessibility of this exposed disulfide may help couple MsrB3 activity to other dithiol-disulfide redox events in the secretory pathway.
-
(2020) Cell. 183, 3, p. 717-729.e16 Abstract[All authors]
The respiratory and intestinal tracts are exposed to physical and biological hazards accompanying the intake of air and food. Likewise, the vasculature is threatened by inflammation and trauma. Mucin glycoproteins and the related von Willebrand factor guard the vulnerable cell layers in these diverse systems. Colon mucins additionally house and feed the gut microbiome. Here, we present an integrated structural analysis of the intestinal mucin MUC2. Our findings reveal the shared mechanism by which complex macromolecules responsible for blood clotting, mucociliary clearance, and the intestinal mucosal barrier form protective polymers and hydrogels. Specifically, cryo-electron microscopy and crystal structures show how disulfide-rich bridges and pH-tunable interfaces control successive assembly steps in the endoplasmic reticulum and Golgi apparatus. Remarkably, a densely O-glycosylated mucin domain performs an organizational role in MUC2. The mucin assembly mechanism and its adaptation for hemostasis provide the foundation for rational manipulation of barrier function and coagulation.
-
(2020) Science Advances. 6, 14, eaay9572. Abstract[All authors]
The endoplasmic reticulum (ER) is a highly dynamic network of membranes. Here, we combine live-cell microscopy with in situ cryo-electron tomography to directly visualize ER dynamics in several secretory cell types including pancreatic beta-cells and neurons under near-native conditions. Using these imaging approaches, we identify a novel, mobile form of ER, ribosome-associated vesicles (RAVs), found primarily in the cell periphery, which is conserved across different cell types and species. We show that RAVs exist as distinct, highly dynamic structures separate from the intact ER reticular architecture that interact with mitochondria via direct intermembrane contacts. These findings describe a new ER subcompartment within cells.
-
(2020) Oncotarget. 11, 4, p. 386-398 Abstract
Extracellular matrix (ECM) plays an important role in tumor development and dissemination, but few points of therapeutic intervention targeting ECM of the tumor microenvironment have been exploited to date. Recent observations suggest that the enzymatic introduction of disulfide bond cross-links into the ECM may be modulated to affect cancer progression. Specifically, the disulfide bond-forming activity of the enzyme Quiescin sulfhydryl oxidase 1 (QSOX1) is required by fibroblasts to assemble ECM components for adhesion and migration of cancer cells. Based on this finding and the increased QSOX1 expression in the stroma of aggressive breast carcinomas, we developed monoclonal antibody inhibitors with the aim of preventing QSOX1 from participating in pro-metastatic ECM remodeling. Here we show that QSOX1 inhibitory antibodies decreased tumor growth and metastasis in murine cancer models and had added benefits when provided together with chemotherapy. Mechanistically, the inhibitors dampened stromal participation in tumor development, as the tumors of treated animals showed fewer myofibroblasts and poorer ECM organization. Thus, our findings demonstrate that specifically targeting excess stromal QSOX1 secreted in response to tumor-cell signaling provides a means to modulate the tumor microenvironment and may complement other therapeutic approaches in cancer.
2019
-
(2019) Journal of Molecular Biology. 431, 19, p. 3740-3752 Abstract
The mucin 2 glycoprotein assembles into a complex hydrogel that protects intestinal epithelia and houses the gut microbiome. A major step in mucin 2 assembly is further multimerization of preformed mucin dimers, thought to produce a honeycomb-like arrangement upon hydrogel expansion. Important open questions are how multiple mucin 2 dimers become covalently linked to one another and how mucin 2 multimerization compares with analogous processes in related polymers such as respiratory tract mucins and the hemostasis protein von Willebrand factor. Here we report the x-ray crystal structure of the mucin 2 multimerization module, found to form a dimer linked by two intersubunit disulfide bonds. The dimer structure calls into question the current model for intestinal mucin assembly, which proposes disulfide-mediated trimerization of the same module. Key residues making interactions across the dimer interface are highly conserved in intestinal mucin orthologs, supporting the physiological relevance of the observed quaternary structure. With knowledge of the interface residues, it can be demonstrated that many of these amino acids are also present in other mucins and in von Willebrand factor, further indicating that the stable dimer arrangement reported herein is likely to be shared across this functionally broad protein family. The mucin 2 module structure thus reveals the manner by which both mucins and von Willebrand factor polymerize, drawing deep structural parallels between macromolecular assemblies critical to mucosal epithelia and the vasculature.
-
(2019) PLoS Computational Biology. 15, 8, e1007207. Abstract
Antibodies developed for research and clinical applications may exhibit suboptimal stability, expressibility, or affinity. Existing optimization strategies focus on surface mutations, whereas natural affinity maturation also introduces mutations in the antibody core, simultaneously improving stability and affinity. To systematically map the mutational tolerance of an antibody variable fragment (Fv), we performed yeast display and applied deep mutational scanning to an anti-lysozyme antibody and found that many of the affinity-enhancing mutations clustered at the variable light-heavy chain interface, within the antibody core. Rosetta design combined enhancing mutations, yielding a variant with tenfold higher affinity and substantially improved stability. To make this approach broadly accessible, we developed AbLIFT, an automated web server that designs multipoint core mutations to improve contacts between specific Fv light and heavy chains (http://AbLIFT.weizmann.ac.il). We applied AbLIFT to two unrelated antibodies targeting the human antigens VEGF and QSOX1. Strikingly, the designs improved stability, affinity, and expression yields. The results provide proof-of-principle for bypassing laborious cycles of antibody engineering through automated computational affinity and stability design.
[All authors] -
(2019) EMBO Journal. 38, 15, 102743. Abstract
ATF6 is a major signal transducer for cellular reprogramming in response to protein mis-folding in the endoplasmic reticulum. However, the mechanism by which ATF6 senses unfolded proteins and becomes activated is not yet known. In this issue of The EMBO Journal, Oka et al show that ERp18, a single-domain member of the protein disulfide isomerase family, interacts preferentially with ATF6 under stress conditions and regulates ATF6 transport to the Golgi apparatus. Furthermore, ERp18 impacts the ATF6 cleavage product generated in the Golgi, ultimately determining whether or not ATF6 becomes a functional transcription factor and induces the unfolded protein response.
-
(2019) Protein Science. 28, 1, p. 228-238 Abstract
The thioredoxin superfamily has expanded and diverged extensively throughout evolution such that distant members no longer show appreciable sequence homology. Nevertheless, redox-active thioredoxin-fold proteins functioning in diverse physiological contexts often share canonical amino acids near the active-site (di-)cysteine motif. Quiescin sulfhydryl oxidase 1 (QSOX1), a catalyst of disulfide bond formation secreted by fibroblasts, is a multi-domain thioredoxin superfamily enzyme with certain similarities to the protein disulfide isomerase (PDI) enzymes. Among other potential functions, QSOX1 supports extracellular matrix assembly in fibroblast cultures. We introduced mutations at a cis-proline in QSOX1 that is conserved across the thioredoxin superfamily and was previously observed to modulate redox interactions of the bacterial enzyme DsbA. The resulting QSOX1 variants showed a striking detrimental effect when added exogenously to fibroblasts: they severely disrupted the extracellular matrix and cell adhesion, even in the presence of naturally secreted, wild-type QSOX1. The specificity of this phenomenon for particular QSOX1 mutants inspired an investigation of the effects of mutation on catalytic and redox properties. For a series of QSOX1 mutants, the detrimental effect correlated with the redox potential of the first redox-active site, and an X-ray crystal structure of one of the mutants revealed the reorganization of the cis-proline loop caused by the mutations. Due to the conservation of the mutated residues across the PDI family and beyond, insights obtained in this study may be broadly applicable to a variety of physiologically important redox-active enzymes. Impact statement We show that mutation of a conserved cis-proline amino acid, analogous to a mutation used to trap substrates of a bacterial disulfide catalyst, has a dramatic effect on the physiological function of the mammalian disulfide catalyst QSOX1. As the active-site region of QSOX1 is shared with the large family of protein disulfide isomerases in humans, the effects of such mutations on redox properties, enzymatic activity, and biological targeting may be relevant across the family.
-
(2019) Journal of Structural Biology: X. 1, 100002. Abstract
Cells and extracellular matrix (ECM) are mutually interdependent: cells guide self-assembly of ECM precursors, and the resulting ECM architecture supports and instructs cells. Though bidirectional signaling between ECM and cells is fundamental to cell biology, it is challenging to gain high-resolution structural information on cellular responses to the matrix microenvironment. Here we used cryo-scanning transmission electron tomography (CSTET) to reveal the nanometer- to micron-scale organization of major fibroblast ECM components in a native-like context, while simultaneously visualizing internal cell ultrastructure including organelles and cytoskeleton. In addition to extending current models for collagen VI fibril organization, three-dimensional views of thick cell regions and surrounding matrix showed how ECM networks impact the structures and dynamics of intracellular organelles and how cells remodel ECM. Collagen VI and fibronectin were seen to distribute in fundamentally different ways in the cell microenvironment and perform distinct roles in supporting and interacting with cells. This work demonstrates that CSTET provides a new perspective for the study of ECM in cell biology, highlighting labeled extracellular elements against a backdrop of unlabeled but morphologically identifiable cellular features with nanometer resolution detail.
2018
-
(2018) Biochemistry. 57, 32, p. 4776-4787 Abstract
Many mutations that cause familial hypercholesterolemia localize to ligand-binding domain 5 (LA5) of the low-density lipoprotein receptor, motivating investigation of the folding and misfolding of this small, disulfide-rich, calcium-binding domain. LA5 folding is known to involve non-native disulfide isomers, yet these folding intermediates have not been structurally characterized. To provide insight into these intermediates, we used nuclear magnetic resonance (NMR) to follow LA5 folding in real time. We demonstrate that misfolded or partially folded disulfide intermediates are indistinguishable from the unfolded state when focusing on the backbone NMR signals, which provide information on the formation of only the final, native state. However,
13C labeling of cysteine side chains differentiated transient intermediates from the unfolded and native states and reported on disulfide bond formation in real time. The cysteine pairings in a dominant intermediate were identified using
13C-edited three-dimensional NMR, and coarse-grained molecular dynamics simulations were used to investigate the preference of this disulfide set over other non-native arrangements. The transient population of LA5 species with particular non-native cysteine connectitivies during folding supports the conclusion that cysteine pairing is not random and that there is a bias toward certain structural ensembles during the folding process, even prior to the binding of calcium. -
(2018) Glycobiology. 28, 8, p. 580-591 Abstract
Quiescin sulfhydryl oxidase 1 (QSOX1) catalyzes the formation of disulfide bonds in protein substrates. Unlike other enzymes with related activities, which are commonly found in the endoplasmic reticulum, QSOX1 is localized to the Golgi apparatus or secreted. QSOX1 is upregulated in quiescent fibroblast cells and secreted into the extracellular environment, where it contributes to extracellular matrix assembly. QSOX1 is also upregulated in adenocarcinomas, though the extent to which it is secreted in this context is currently unknown. To achieve a better understanding of factors that dictate QSOX1 localization and function, we aimed to determine how post-translational modifications affect QSOX1 trafficking and activity. We found a highly conserved N-linked glycosylation site to be required for QSOX1 secretion from fibroblasts and other cell types. Notably, QSOX1 lacking a glycan at this site arrives at the Golgi, suggesting that it passes endoplasmic reticulum quality control but is not further transported to the cell surface for secretion. The QSOX1 transmembrane segment is dispensable for Golgi localization and secretion, as fully luminal and transmembrane variants displayed the same trafficking behavior. This study provides a key example of the effect of glycosylation on Golgi exit and contributes to an understanding of late secretory sorting and quality control.
-
(2018) Chemical Reviews. 118, 3, p. 293-322 Abstract
Cysteine thiols are among the most reactive functional groups in proteins, and their pairing in disulfide linkages is a common post-translational modification in proteins entering the secretory pathway. This modest amino acid alteration, the mere removal of a pair of hydrogen atoms from juxtaposed cysteine residues, contrasts with the substantial changes that characterize most other post-translational reactions. However, the wide variety of proteins that contain disulfides, the profound impact of cross-linking on the behavior of the protein polymer, the numerous and diverse players in intracellular pathways for disulfide formation, and the distinct biological settings in which disulfide bond formation can take place belie the simplicity of the process. Here we lay the groundwork for appreciating the mechanisms and consequences of disulfide bond formation in vivo by reviewing chemical principles underlying cysteine pairing and oxidation. We then show how enzymes tune redox-active cofactors and recruit oxidants to improve the specificity and efficiency of disulfide formation. Finally, we discuss disulfide bond formation in a cellular context and identify important principles that contribute to productive thiol oxidation in complex, crowded, dynamic environments.
2017
-
(2017) eLife. 6, e29929. Abstract
The entry of calcium into mitochondria is central to metabolism, inter-organelle communication, and cell life/death decisions. Long-sought transporters involved in mitochondrial calcium influx and efflux have recently been identified. To obtain a unified picture of mitochondrial calcium utilization, a parallel advance in understanding the forms and quantities of mitochondrial calcium stores is needed. We present here the direct 3D visualization of mitochondrial calcium in intact mammalian cells using cryo-scanning transmission electron tomography (CSTET). Amorphous solid granules containing calcium and phosphorus were pervasive in the mitochondrial matrices of a variety of mammalian cell types. Analysis based on quantitative electron scattering revealed that these repositories are equivalent to molar concentrations of dissolved ions. These results demonstrate conclusively that calcium buffering in the mitochondrial matrix in live cells occurs by phase separation, and that solid-phase stores provide a major ion reservoir that can be mobilized for bioenergetics and signaling.
2016
-
(2016) Proceedings of the National Academy of Sciences - PNAS. 113, 47, p. 13384-13389 Abstract
Laminin, an ∼800-kDa heterotrimeric protein, is a major functional component of the extracellular matrix, contributing to tissue development and maintenance. The unique architecture of laminin is not currently amenable to determination at high resolution, as its flexible and narrowsegments complicate both crystallization and single-particle reconstruction by electronmicroscopy. Therefore, we used cross-linking and MS, evaluated using computational methods, to address key questions regarding laminin quaternary structure. This approach was particularly well suited to the ∼750-Å coiled coil that mediates trimer assembly, and our results support revision of the subunit order typically presented in laminin schematics. Furthermore, information on the subunit register in the coiled coil and cross-links to downstream domains provide insights into the self-assembly required for interaction with other extracellular matrix and cell surface proteins.
-
(2016) Free Radical Biology and Medicine. 99, p. 426-435 Abstract
Increased thioredoxin reductase (TrxR) levels in serum were recently identified as possible prognostic markers for human prostate cancer or hepatocellular carcinoma. We had earlier shown that serum levels of TrxR protein are very low in healthy mice, but can in close correlation to alanine aminotransferase (ALT) increase more than 200-fold upon chemically induced liver damage. We also found that enzymatic TrxR activity in serum is counteracted by a yet unidentified oxidase activity in serum. In the present study we found that mice carrying H22 hepatocellular carcinoma tumors present highly increased levels of TrxR in serum, similarly to that reported in human patients. In this case ALT levels did not parallel those of TrxR. We also discovered here that the TrxR-antagonistic oxidase activity in serum is due to the presence of quiescin Q6 sulfhydryl oxidase 1 (QSOX1). We furthermore found that the chemotherapeutic agents cisplatin or auranofin, when given systemically to H22 tumor bearing mice, can further inhibit TrxR activities in serum. The TrxR serum activity was also inhibited by endogenous electrophilic inhibitors, found to increase in tumor-bearing mice and to include protoporphyrin IX (PpIX) and 4-hydroxynonenal (HNE). Thus, hepatocellular carcinoma triggers high levels of serum TrxR that are not paralleled by ALT, and TrxR enzyme activity in serum is counteracted by several different mechanisms. The physiological role of TrxR in serum, if any, as well as its potential value as a prognostic marker for tumor progression, needs to be studied further. (C) 2016 Elsevier Inc. All rights reserved.
-
(2016) Proteins. 84, S1, p. 34-50 Abstract
The Critical Assessment of protein Structure Prediction (CASP) experiment would not have been possible without the prediction targets provided by the experimental structural biology community. In this article, selected crystallographers providing targets for the CASP11 experiment discuss the functional and biological significance of the target proteins, highlight their most interesting structural features, and assess whether these features were correctly reproduced in the predictions submitted to CASP11. Proteins 2016; 84(Suppl 1):3450.
[All authors] -
(2016) Biophysical Journal. 110, 3, p. 23A Abstract
We recently demonstrated that cryo-scanning transmission electron tomography (CSTET) provides tomographic reconstructions of vitrified cells with superior information transfer at high tilts and for thicker specimens than defocus phase contrast (Wolf et al., 2014). In cryoSTEM, there are no image-forming lenses after the electron beam passes through the sample; detection is incoherent and inelastically scattered electrons provide usable contrast information. By obviating the need for zero-loss energy filtration, the STEM modality provides efficient use of electron dose, thereby minimizing specimen damage. Here we demonstrate the use of CSTET for obtaining highly detailed 3D architectures of organelles and macromolecular complexes in unstained, unfixed, and unsectioned cultured fibroblasts while simultaneously collecting analytical information from high-angle, incoherently scattered electrons. As a case in point, cryoSTEM tomograms revealed characteristic patterns of dense deposits sequestered in mitochondria. Energy-dispersive X-ray (EDX) spectroscopy of these deposits revealed calcium and phosphorus. Once the elemental identification was made, the STEM scattering signal could be interpreted quantitatively as a three-dimensional map of mitochondrial calcium deposition. This approach can be extended to identify and map other concentrations of elements in the cell heavier than the pervasive carbon, nitrogen, and oxygen, as we demonstrated for phosphorus in bacterial cells (Wolf et al., 2015). This study provides an example of how imaging with sensitivity to atomic number in whole cells will provide a new dimension in structural cell biology by correlating elemental composition to organelle morphology.
-
(2016) Protein engineering, design & selection : PEDS. 29, 4, p. 135-147 Abstract
The secreted disulfide catalyst Quiescin sulfhydryl oxidase-1 (QSOX1) affects extracellular matrix organization and is overexpressed in various adenocarcinomas and associated stroma. Inhibition of extracellular human QSOX1 by a monoclonal antibody decreased tumor cell migration in a cell co-culture model and hence may have therapeutic potential. However, the species specificity of the QSOX1 monoclonal antibody has been a setback in assessing its utility as an anti-metastatic agent in vivo, a common problem in the antibody therapy industry. We therefore used structurally guided engineering to expand the antibody species specificity, improving its affinity toward mouse QSOX1 by at least four orders of magnitude. A crystal structure of the re-engineered variant, complexed with its mouse antigen, revealed that the antibody accomplishes dual-species targeting through altered contacts between its heavy and light chains, plus replacement of bulky aromatics by flexible side chains and versatile water-bridged polar interactions. In parallel, we produced a surrogate antibody targeting mouse QSOX1 that exhibits a new QSOX1 inhibition mode. This set of three QSOX1 inhibitory antibodies is compatible with various mouse models for pre-clinical trials and biotechnological applications. In this study we provide insights into structural blocks to cross-reactivity and set up guideposts for successful antibody design and re-engineering.
-
(2016) Free Radical Biology and Medicine. 99, p. 426–435
2015
-
(2015) Nature Communications. 6, 8624. Abstract
The ability to query enzyme molecules individually is transforming our view of catalytic mechanisms. Quiescin sulfhydryl oxidase (QSOX) is a multidomain catalyst of disulfide-bond formation that relays electrons from substrate cysteines through two redox-active sites to molecular oxygen. The chemical steps in electron transfer have been delineated, but the conformational changes accompanying these steps are poorly characterized. Here we use single-molecule Förster resonance energy transfer (smFRET) to probe QSOX conformation in resting and cycling enzyme populations. We report the discovery of unanticipated roles for conformational changes in QSOX beyond mediating electron transfer between redox-active sites. In particular, a state of the enzyme not previously postulated or experimentally detected is shown to gate, via a conformational transition, the entrance into a sub-cycle within an expanded QSOX kinetic scheme. By tightly constraining mechanistic models, smFRET data can reveal the coupling between conformational and chemical transitions in complex enzymatic cycles.
-
(2015) FEBS Journal. 282, 14, p. 2746-2757 Abstract
The ∼ 800 kDa laminin heterotrimer forms a distinctive cross-shaped structure that further self-assembles into networks within the extracellular matrix. The domains at the laminin chain termini, which engage in network formation and cell-surface interaction, are well understood both structurally and functionally. By contrast, the structures and roles of additional domains embedded within the limbs of the laminin cross have remained obscure. Here, we report the X-ray crystal structure, determined to 1.2 Å resolution, of the human laminin α2 subunit L4b domain, site of an inframe deletion mutation associated with mild congenital muscular dystrophy. The α2 L4b domain is an irregular β-sandwich with many short and broken strands linked by extended loops. The most similar known structures are the carbohydrate-binding domains of bacterial cellulases, the ephrin-binding domain of ephrin receptors, and MAM adhesion domains in various other eukaryotic cell-surface proteins. This similarity to mammalian adhesion modules, which was not predicted on the basis of amino acid sequence alone due to lack of detectable homology, suggests that laminin internal domains evolved from a progenitor adhesion molecule and may retain a role in cell adhesion in the context of the laminin trimer.
-
(2015) AIMS Biophysics. 2, 3, p. 259-273 Abstract
Cryo-tomography of intact, vitrified cells provides a three dimensional view of their structure and organization in a snapshot of the living state. Lacking heavy metal stains, tilt series images are typically produced by defocus phase contrast. Recently, a number of other methods have been introduced for 3D cryo-imaging. These include phase plate imaging, soft X-ray tomography, serial surface imaging using the focused ion beam-scanning electron microscope, and cryo-STEM tomography (CSTET). Here we explain the basis of the STEM setup and demonstrate the capabilities of CSTET to study unfixed, fully hydrated mammalian cells. Numerous cellular features are recognized in CSTET reconstructions, including membranes, vesicles, cytoskeleton, extracellular matrix, coated pits, and ribosomes. STEM signal acquisition configuration is more flexible than defocus phase contrast, and it imposes a much less severe spatial filter on the original images. Because low spatial frequency information is retained, the STEM tomographic reconstruction more faithfully represents the mass density distribution in the specimen.
2014
-
(2014) PLoS ONE. 9, 12, e113431. Abstract
The widespread thioredoxin superfamily enzymes typically share the following features: a characteristic α-β fold, the presence of a Cys-X-X-Cys (or Cys-X-X-Ser) redox-active motif, and a proline in the cis configuration abutting the redox-active site in the tertiary structure. The Cys-X-X-Cys motif is at the solvent-exposed amino terminus of an a-helix, allowing the first cysteine to engage in nucleophilic attack on substrates, or substrates to attack the Cys-X-X-Cys disulfide, depending on whether the enzyme functions to reduce, isomerize, or oxidize its targets. We report here the X-ray crystal structure of an enzyme that breaks many of our assumptions regarding the sequence-structure relationship of thioredoxin superfamily proteins. The yeast Protein Disulfide Isomerase family member Eps1p has Cys-X-X-Cys motifs and proline residues at the appropriate primary structural positions in its first two predicted thioredoxin-fold domains. However, crystal structures show that the Cys-X-X-Cys of the second domain is buried and that the adjacent proline is in the trans, rather than the cis isomer. In these configurations, neither the "active-site" disulfide nor the backbone carbonyl preceding the proline is available to interact with substrate. The Eps1p structures thus expand the documented diversity of the PDI oxidoreductase family and demonstrate that conserved sequence motifs in common folds do not guarantee structural or functional conservation.
-
-
(2014) Protein Science. 23, 8, p. 1102-1112 Abstract
Thioredoxin superfamily proteins introduce disulfide bonds into substrates, catalyze the removal of disulfides, and operate in electron relays. These functions rely on one or more dithiol/ disulfide exchange reactions. The flavoenzyme quiescin sulfhydryl oxidase (QSOX), a catalyst of disulfide bond formation with an interdomain electron transfer step in its catalytic cycle, provides a unique opportunity for exploring the structural environment of enzymatic dithiol/disulfide exchange. Wild-type Rattus norvegicus QSOX1 (RnQSOX1) was crystallized in a conformation that juxtaposes the two redox-active di-cysteine motifs in the enzyme, presenting the entire electron-transfer pathway and proton-transfer participants in their native configurations. As such a state cannot generally be enriched and stabilized for analysis, RnQSOX1 gives unprecedented insight into the functional group environments of the four cysteines involved in dithiol/disulfide exchange and provides the framework for analysis of the energetics of electron transfer in the presence of the bound flavin adenine dinucleotide cofactor. Hybrid quantum mechanics/molecular mechanics (QM/MM) free energy simulations based on the X-ray crystal structure suggest that formation of the interdomain disulfide intermediate is highly favorable and secures the flexible enzyme in a state from which further electron transfer via the flavin can occur.
-
(2014) Proteins-Structure Function And Bioinformatics. 82, SUPPL.2, p. 26-42 Abstract[All authors]
For the last two decades, CASP has assessed the state of the art in techniques for protein structure prediction and identified areas which required further development. CASP would not have been possible without the prediction targets provided by the experimental structural biology community. In the latest experiment, CASP10, more than 100 structures were suggested as prediction targets, some of which appeared to be extraordinarily difficult for modeling. In this article, authors of some of the most challenging targets discuss which specific scientific question motivated the experimental structure determination of the target protein, which structural features were especially interesting from a structural or functional perspective, and to what extent these features were correctly reproduced in the predictions submitted to CASP10. Specifically, the following targets will be presented: the acid-gated urea channel, a difficult to predict transmembrane protein from the important human pathogen Helicobacter pylori; the structure of human interleukin (IL)-34, a recently discovered helical cytokine; the structure of a functionally uncharacterized enzyme OrfY from Thermoproteus tenax formed by a gene duplication and a novel fold; an ORFan domain of mimivirus sulfhydryl oxidase R596; the fiber protein gene product 17 from bacteriophage T7; the bacteriophage CBA-120 tailspike protein; a virus coat protein from metagenomic samples of the marine environment; and finally, an unprecedented class of structure prediction targets based on engineered disulfide-rich small proteins.
2013
-
(2013) Journal of Molecular Biology. 425, 22, p. 4366-4378 Abstract
Quiescin sulfhydryl oxidase 1 (QSOX1) is a catalyst of disulfide bond formation that undergoes regulated secretion from fibroblasts and is over-produced in adenocarcinomas and other cancers. We have recently shown that QSOX1 is required for incorporation of particular laminin isoforms into the extracellular matrix (ECM) of cultured fibroblasts and, as a consequence, for tumor cell adhesion to and penetration of the ECM. The known role of laminins in integrin-mediated cell survival and motility suggests that controlling QSOX1 activity may provide a novel means of combating metastatic disease. With this motivation, we developed a monoclonal antibody that inhibits the activity of human QSOX1. Here, we present the biochemical and structural characterization of this antibody and demonstrate that it is a tight-binding inhibitor that blocks one of the redox-active sites in the enzyme, but not the site at which de novo disulfides are generated catalytically. Sulfhydryl oxidase activity is thus prevented without direct binding of the sulfhydryl oxidase domain, confirming the model for the interdomain QSOX1 electron transfer mechanism originally surmised based on mutagenesis and protein dissection. In addition, we developed a single-chain variant of the antibody and show that it is a potent QSOX1 inhibitor. The QSOX1 inhibitory antibody will be a valuable tool in studying the role of ECM composition and architecture in cell migration, and the recombinant version may be further developed for potential therapeutic applications based on manipulation of the tumor microenvironment.
-
(2013) Molecular Cell. 51, 3, p. 281-282 Abstract
In this issue, Lee etal. (2013) exhibit methionine sulfoxidation in a new light. By bringing together two antagonistic enzymes affecting methionine redox state, the authors demonstrate that methionine oxidation constitutes a reversible, posttranslational regulatory mechanism, akin to protein phosphorylation.
-
(2013) Science. 341, 6141, p. 74-76 Abstract
Disulfide bond formation in secretory proteins occurs primarily in the endoplasmic reticulum (ER), where multiple enzyme families catalyze cysteine cross-linking. Quiescin sulfhydryl oxidase 1 (QSOX1) is an atypical disulfide catalyst, localized to the Golgi apparatus or secreted from cells. We examined the physiological function for extracellular catalysis of de novo disulfide bond formation by QSOX1. QSOX1 activity was required for incorporation of laminin into the extracellular matrix (ECM) synthesized by fibroblasts, and ECM produced without QSOX1 was defective in supporting cell-matrix adhesion. We developed an inhibitory monoclonal antibody against QSOX1 that could modulate ECM properties and undermine cell migration.
-
(2013) BMC Evolutionary Biology. 13, 1, 70. Abstract
Background: The enzyme family Quiescin Sulfhydryl Oxidase (QSOX) is defined by the presence of an amino-terminal thioredoxin-fold (Trx) domain and a carboxy-terminal Erv family sulfhydryl oxidase domain. QSOX enzymes, which generate disulfide bonds and transfer them to substrate proteins, are present in a wide variety of eukaryotic species including metazoans and plants, but are absent from fungi. Plant and animal QSOXs differ in their active-site amino acid sequences and content of non-catalytic domains. The question arises, therefore, whether the Trx-Erv fusion has the same mechanistic significance in all QSOX enzymes, and whether shared features distinguish the functional domains of QSOX from other instances in which these domains occur independently. Through a study of QSOX phylogeny and an analysis of QSOX sequence diversity in light of recently determined three-dimensional structures, we sought insight into the origin and evolution of this multi-domain redox alliance. Results: An updated collection of QSOX enzymes was used to confirm and refine the differences in domain composition and active-site sequence motif patterns of QSOXs belonging to various eukaryotic phyla. Beyond the expected phylogenetic distinction of animal and plant QSOX enzymes, trees based on individual redox-active QSOX domains show a particular distinction of the Trx domain early in plant evolution. A comparison of QSOX domains with Trx and Erv domains from outside the QSOX family revealed several sequence and structural features that clearly differentiate QSOXs from other enzymes containing either of these domains. Notably, these features, present in QSOXs of various phyla, localize to the interface between the Trx and Erv domains observed in structures of QSOX that model interdomain redox communication. Conclusions: The infrastructure for interdomain electron relay, previously identified for animal and parasite QSOXs, is found broadly across the QSOX family, including the plant enzymes. We conclude that the conserved three-dimensional framework of the QSOX catalytic domains accommodates lineage-specific differences and paralog diversification in the amino acid residues surrounding the redox-active cysteines. Our findings indicate that QSOX enzymes are characterized not just by the presence of the two defining domain folds but also by features that promote coordinated activity.
2012
-
(2012) FEBS Letters. 586, 23, p. 4119-4125 Abstract
Quiescin Sulfhydryl Oxidase (QSOX), a catalyst of disulfide bond formation, is found in both plants and animals. Mammalian, avian, and trypanosomal QSOX enzymes have been studied in detail, but plant QSOX has yet to be characterized. Differences between plant and animal QSOXs in domain composition and active-site sequences raise the question of whether these QSOXs function by the same mechanism. We demonstrate that Arabidopsis thaliana QSOX produced in bacteria is folded and functional as a sulfhydryl oxidase but does not exhibit the interdomain electron transfer observed for its animal counterpart. Based on this finding, further exploration into the respective roles of the redox-active sites in plant QSOX and the reason for their concatenation is warranted.
-
(2012) PLoS ONE. 7, 11, e50649. Abstract
The mimivirus genome contains many genes that lack homologs in the sequence database and are thus known as ORFans. In addition, mimivirus genes that encode proteins belonging to known fold families are in some cases fused to domain-sized segments that cannot be classified. One such ORFan region is present in the mimivirus enzyme R596, a member of the Erv family of sulfhydryl oxidases. We determined the structure of a variant of full-length R596 and observed that the carboxy-terminal region of R596 assumes a folded, compact domain, demonstrating that these ORFan segments can be stable structural units. Moreover, the R596 ORFan domain fold is novel, hinting at the potential wealth of protein structural innovation yet to be discovered in large double-stranded DNA viruses. In the context of the R596 dimer, the ORFan domain contributes to formation of a broad cleft enriched with exposed aromatic groups and basic side chains, which may function in binding target proteins or localization of the enzyme within the virus factory or virions. Finally, we find evidence for an intermolecular dithiol/disulfide relay within the mimivirus R596 dimer, the first such extended, intersubunit redox-active site identified in a viral sulfhydryl oxidase.
-
(2012) Nature. 488, 7411, p. 414-418 Abstract
Protein stability, assembly, localization and regulation often depend on the formation of disulphide crosslinks between cysteine side chains. Enzymes known as sulphydryl oxidases catalyse de novo disulphide formation and initiate intra-and intermolecular dithiol/disulphide relays to deliver the disulphides to substrate proteins. Quiescin sulphydryl oxidase (QSOX) is a unique, multi-domain disulphide catalyst that is localized primarily to the Golgi apparatus and secreted fluids and has attracted attention owing to its overproduction in tumours. In addition to its physiological importance, QSOX is a mechanistically intriguing enzyme, encompassing functions typically carried out by a series of proteins in other disulphide-formation pathways. How disulphides are relayed through the multiple redox-active sites of QSOX and whether there is a functional benefit to concatenating these sites on a single polypeptide are open questions. Here we present the first crystal structure of an intact QSOX enzyme, derived from a trypanosome parasite. Notably, sequential sites in the disulphide relay were found more than 40Å apart in this structure, too far for direct disulphide transfer. To resolve this puzzle, we trapped and crystallized an intermediate in the disulphide hand-off, which showed a 165 ° domain rotation relative to the original structure, bringing the two active sites within disulphide-bonding distance. The comparable structure of a mammalian QSOX enzyme, also presented here, shows further biochemical features that facilitate disulphide transfer in metazoan orthologues. Finally, we quantified the contribution of concatenation to QSOX activity, providing general lessons for the understanding of multi-domain enzymes and the design of new catalytic relays.
[All authors] -
(2012) Annual Review of Biophysics. 41, 1, p. 63-79 Abstract
It has been known for many decades that cell surface, soluble-secreted, and extracellular matrix proteins are generally rich in disulfide bonds, but only more recently has the functional diversity of disulfide bonding in extracellular proteins been appreciated. In addition to the classic mechanisms by which disulfide bonds enhance protein thermodynamic stability, disulfides in certain configurations contribute particular mechanical properties to proteins that sense and respond to tensile forces. Disulfides may help warp protein folds for the evolution of new functions, or they may fasten aggregation-prone flaps of polypeptide to protein surfaces to prevent fibrilization or oligomerization. Disulfides can also be used to package and secure macromolecular cargo for intercellular transport. A series of case studies illustrating diverse biophysical roles of disulfide bonding are reviewed, with a focus on proteins functioning in the extracellular environment.
2011
-
(2011) Journal of Virology. 85, 18, p. 9406-9413 Abstract
Genomes of nucleocytoplasmic large DNA viruses (NCLDVs) encode enzymes that catalyze the formation of disulfide bonds between cysteine amino acid residues in proteins, a function essential for the proper assembly and propagation of NCLDV virions. Recently, a catalyst of disulfide formation was identified in baculoviruses, a group of large double-stranded DNA viruses considered phylogenetically distinct from NCLDVs. The NCLDV and baculovirus disulfide catalysts are flavin adenine dinucleotide (FAD)-binding sulfhydryl oxidases related to the cellular Erv enzyme family, but the baculovirus enzyme, the product of the Ac92 gene in Autographa californica multiple nucleopolyhedrovirus (AcMNPV), is highly divergent at the amino acid sequence level. The crystal structure of the Ac92 protein presented here shows a configuration of the active-site cysteine residues and bound cofactor similar to that observed in other Erv sulfhydryl oxidases. However, Ac92 has a complex quaternary structural arrangement not previously seen in cellular or viral enzymes of this family. This novel assembly comprises a dimer of pseudodimers with a striking 40-degree kink in the interface helix between subunits. The diversification of the Erv sulfhydryl oxidase enzymes in large double-stranded DNA viruses exemplifies the extreme degree to which these viruses can push the boundaries of protein family folds.
-
(2011) Nature. 473, 7348, p. 540-543 Abstract[All authors]
Molecular replacement procedures1-4, which search for placements of a starting model within the crystallographic unit cell that best account for the measured diffraction amplitudes, followed by automatic chain tracing methods5-8, have allowed the rapid solution of large numbers of protein crystal structures. Despite extensive work9-14, molecular replacement or the subsequent rebuilding usually fail with more divergent starting models based on remote homologues with less than 30% sequence identity. Here we show that this limitation can be substantially reduced by combining algorithms for protein structure modelling with those developed for crystallographic structure determination. An approach integrating Rosetta structure modelling with Autobuild chain tracing yielded high-resolution structures for 8 of 13 X-ray diffraction data sets that could not be solved in the laboratories of expert crystallographers and that remained unsolved after application of an extensive array of alternative approaches. We estimate that the new method should allow rapid structure determination without experimental phase information for over half the cases where current methods fail, given diffraction data sets of better than 3.2 Å resolution, four or fewer copies in the asymmetric unit, and the availability of structures of homologous proteins with >20% sequence identity.
2010
-
(2010) Molecular Cell. 40, 5, p. 685-686 Abstract
In this issue of Molecular Cell, Ron and colleagues (Zito et al., 2010b) show that an enzyme responsible for cleaning up hydrogen peroxide in the endoplasmic reticulum can contribute productively to disulfide bond formation.
-
(2010) Antioxidants & Redox Signaling. 13, 8, p. 1261-1271 Abstract
Proteins that have evolved to contain stabilizing disulfide bonds generally fold in a membrane-delimited compartment in the cell [i.e., the endoplasmic reticulum (ER) or the mitochondrial intermembrane space (IMS)]. These compartments contain sulfhydryl oxidase enzymes that catalyze the pairing and oxidation of cysteine residues. In contrast, most proteins in a healthy cytosol are maintained in reduced form through surveillance by NADPH-dependent reductases and the lack of sulfhydryl oxidases. Nevertheless, one of the core functionalities that unify the broad and diverse set of nucleocytoplasmic large DNA viruses (NCLDVs) is the ability to catalyze disulfide formation in the cytosol. The substrates of this activity are proteins that contribute to the assembly, structure, and infectivity of the virions. If the last common ancestor of NCLDVs was present during eukaryogenesis as has been proposed, it is interesting to speculate that viral disulfide bond formation pathways may have predated oxidative protein folding in intracellular organelles.
-
(2010) Protein Science. 19, 10, p. 1863-1876 Abstract
Ero1p is the primary catalyst of disulfide bond formation in the yeast endoplasmic reticulum (ER). Ero1p contains a pair of essential disulfide bonds that participate directly in the electron transfer pathway from substrate thiol groups to oxygen. Remarkably, elimination of certain other Ero1p disulfides by mutation enhances enzyme activity. In particular, the C150A/C295A Ero1p mutant exhibits increased thiol oxidation in vitro and in vivo and interferes with redox homeostasis in yeast cells by hyperoxidizing the ER. Inhibitory disulfides of Ero1p are thus important for enzyme regulation. To visualize the differences between de-regulated and wild-type Ero1p, we determined the crystal structure of Ero1p C150A/C295A. The structure revealed local changes compared to the wild-type enzyme around the sites of mutation, but no conformational transitions within 25 Å of the active site were observed. To determine how the C150-C295 disulfide nonetheless participates in redox regulation of Ero1p, we analyzed using mass spectrometry the changes in Ero1p disulfide connectivity as a function of time after encounter with reducing substrates. We found that the C150-C295 disulfide sets a physiologically appropriate threshold for enzyme activation by guarding a key neighboring disulfide from reduction. This study illustrates the diverse and interconnected roles that disulfides can play in redox regulation of protein activity.
-
(2010) Journal of Molecular Biology. 402, 2, p. 388-398 Abstract
The highly conserved Rag family GTPases have a role in reporting amino acid availability to the TOR (target of rapamycin) signaling complex, which regulates cell growth and metabolism in response to environmental cues. The yeast Rag proteins Gtr1p and Gtr2p were shown in multiple independent studies to interact with the membrane-associated proteins Gse1p (Ego3p) and Gse2p (Ego1p). However, mammalian orthologs of Gse1p and Gse2p could not be identified. We determined the crystal structure of Gse1p and found it to match the fold of two mammalian proteins, MP1 (mitogen-activated protein kinase scaffold protein 1) and p14, which form a heterodimeric complex that had been assigned a scaffolding function in mitogen-activated protein kinase pathways. The significance of this structural similarity is validated by the recent identification of a physical and functional association between mammalian Rag proteins and MP1/p14. Together, these findings reveal that key components of the TOR signaling pathway are structurally conserved between yeast and mammals, despite divergence of sequence to a degree that thwarts detection through simple homology searches.
-
(2010) Journal of Biological Chemistry. 285, 27, p. 20993-21003 Abstract
Endoplasmic reticulum oxidation 1 (ERO1) is a conserved eukaryotic flavin adenine nucleotide-containing enzyme that promotes disulfide bond formation by accepting electrons from reduced protein disulfide isomerase (PDI) and passing them on to molecular oxygen. Although disulfide bond formation is an essential process, recent experiments suggest a surprisingly broad tolerance to genetic manipulations that attenuate the rate of disulfide bond formation and that a hyperoxidizing ER may place stressed cells at a disadvantage. In this study, we report on the development of a high throughput in vitro assay for mammalian ERO1α activity and its application to identify small molecule inhibitors. The inhibitor EN460 (IC50, 1.9 μM) interacts selectively with the reduced, active form of ERO1α and prevents its reoxidation. Despite rapid and promiscuous reactivity with thiolates, EN460 exhibits selectivity for ERO1. This selectivity is explained by the rapid reversibility of the reaction of EN460 with unstructured thiols, in contrast to the formation of a stable bond with ERO1α followed by displacement of bound flavin adenine dinucleotide from the active site of the enzyme. Modest concentrations of EN460 and a functionally related inhibitor, QM295, promote signaling in the unfolded protein response and precondition cells against severe ER stress. Together, these observations point to the feasibility of targeting the enzymatic activity of ERO1α with small molecule inhibitors.
-
(2010) Journal of Biological Chemistry. 285, 24, p. 18155-18165 Abstract[All authors]
The sulfhydryl oxidase Ero1 oxidizes protein disulfide isomerase (PDI), which in turn catalyzes disulfide formation in proteins folding in the endoplasmic reticulum (ER). The extent to which other members of the PDI family are oxidized by Ero1 and thus contribute to net disulfide formation in the ER has been an open question. The yeast ER contains four PDI family proteins with at least one potential redox-active cysteine pair. We monitored the direct oxidation of each redox-active site in these proteins by yeast Ero1p in vitro. In this study, we found that the Pdi1p amino-terminal domain was oxidized most rapidly compared with the other oxidoreductase active sites tested, including the Pdi1p carboxyl-terminal domain. This observation is consistent with experiments conducted in yeast cells. In particular, the amino-terminal domain of Pdi1p preferentially formed mixed disulfides with Ero1p in vivo, and we observed synthetic lethality between a temperature-sensitive Ero1p variant and mutant Pdi1p lacking the amino-terminal active-site disulfide. Thus, the amino-terminal domain of yeast Pdi1p is on a preferred pathway for oxidizing the ER thiol pool. Overall, our results provide a rank order for the tendency of yeast ER oxidoreductases to acquire disulfides from Ero1p.
-
(2010) FEBS Letters. 584, 8, p. 1521-1525 Abstract
Quiescin sulfhydryl oxidase (QSOX) catalyzes formation of disulfide bonds between cysteine residues in substrate proteins. Human QSOX1 is a multi-domain, monomeric enzyme containing a module related to the single-domain sulfhydryl oxidases of the Erv family. A partial QSOX1 crystal structure reveals a single-chain pseudo-dimer mimicking the quaternary structure of Erv enzymes. However, one pseudo-dimer " subunit" has lost its cofactor and catalytic activity. In QSOX evolution, a further concatenation to a member of the protein disulfide isomerase family resulted in an enzyme capable of both disulfide formation and efficient transfer to substrate proteins.
2009
-
(2009) Journal of Molecular Biology. 391, 4, p. 758-768 Abstract
Large double-stranded DNA viruses, including poxviruses and mimiviruses, encode enzymes to catalyze the formation of disulfide bonds in viral proteins produced in the cell cytosol, an atypical location for oxidative protein folding. These viral disulfide catalysts belong to a family of sulfhydryl oxidases that are dimers of a small five-helix fold containing a Cys-X-X-Cys motif juxtaposed to a flavin adenine dinucleotide cofactor. We report that the sulfhydryl oxidase pB119L from African swine fever virus (ASFV) uses for self-assembly surface different from that observed in homologs from mammals, plants, and fungi. Within a protein family, different packing interfaces for the same oligomerization state are extremely rare. We find that the alternate dimerization mode seen in ASFV pB119L is not characteristic of all viral sulfhydryl oxidases, as the flavin-binding domain from a mimivirus sulfhydryl oxidase assumes the same dimer structure as the known eukaryotic enzymes. ASFV pB119L demonstrates the potential of large double-stranded DNA viruses, which have faster mutation rates than their hosts and the tendency to incorporate host genes, to pioneer new protein folds and self-assembly modes.
-
(2009) Nature. 459, 7245, p. 371-378 Abstract
Intramembrane proteolysis is increasingly seen as a regulatory step in a range of diverse processes, including development, organelle shaping, metabolism, pathogenicity and degenerative disease. Initial scepticism over the existence of intramembrane proteases was soon replaced by intense exploration of their catalytic mechanisms, substrate specificities, regulation and structures. Crystal structures of metal-dependent and serine intramembrane proteases have revealed active sites embedded in the plane of the membrane but accessible by water, a requirement for hydrolytic reactions. Efforts to understand how these membrane-bound proteases carry out their reactions have started to yield results.
-
(2009) Journal of Biological Chemistry. 284, 4, p. 2098-2105 Abstract
The redox reactivity of the three disulfide bridges and the flavin present in each protomer of the wild-type Arabidopsis thaliana mitochondrial sulfhydryl oxidase (AtErv1) homodimer has been investigated. Pulse radiolytically produced CO2- radical ions were found to reduce the disulfide bridges to yield disulfide radicals, RSS*R-. Rates and absorption changes due to formation or decay of RSS*R- and the flavin quinone, semiquinone, and hydroquinone were measured and analyzed. During the first 100 μs following the pulse, the flavin was reduced to the semiquinone by intramolecular electron transfer from the active site disulfide radical. The semiquinone and the remaining disulfide radicals then reacted by much slower, 40 ms to 40 s, inter-homodimer electron transfer reactions, culminating in reduced flavin and dithiols. The dithiols were then subject to oxidation by enzyme molecules via their intrinsic enzymatic activity, at a rate comparable to the slower intermolecular processes in the 10-s time regime. Mutants of AtErv1 lacking each of the three individual cysteine pairs were studied to determine the involvement of the respective disulfide groups in these reactions. Elimination of the active site disulfide bridge increased the stability of the flavin semiquinone making it a long-lived product. Relevance of these observations to the design and function of the sulfhydryl oxidases is discussed.
2008
-
(2008) Journal of Molecular Biology. 384, 3, p. 631-640 Abstract
Oxidoreductases belonging to the protein disulfide isomerase (PDI) family promote proper disulfide bond formation in substrate proteins in the endoplasmic reticulum. In plants and metazoans, new family members continue to be identified and assigned to various functional niches. PDI-like proteins typically contain tandem thioredoxin-fold domains. The limited information available suggested that the relative orientations of these domains may be quite uniform across the family, and structural models based on this assumption are appearing. However, the X-ray crystal structure of the yeast PDI family protein Mpd1p, described here, demonstrates the radically different domain orientations and surface properties achievable with multiple copies of the thioredoxin fold. A comparison of Mpd1p with yeast Pdi1p expands our perspective on the contexts in which redox-active motifs are presented in the PDI family.
-
(2008) Crystal Growth & Design. 8, 8, p. 2954-2963 Abstract
The fold of the β-helical antifreeze protein from Tenebrio molitor (TmAFP) proved to be surprisingly tolerant of multiple amino acid substitutions, enabling the construction of a panel of mutants displaying grids of single amino acid types in place of the threonines on the ice-binding face. These mutants, maintaining the regularity of amino acid spacing found in the wild-type protein but with different functional groups on the surface, were tested for antifreeze activity by measuring thermal hysteresis and observing ice grown in their presence. We found that no mutant exhibited the dramatic activity of the wild-type version of this hyperactive antifreeze protein. However, mutants containing four valines or tyrosines in place of the threonines in the center of the TmAFP ice-binding face showed residual thermal hysteresis activity and had marked effects on ice crystal morphology. The results are discussed in the context of a two-stage model for the absorption-inhibition mechanism of antifreeze protein binding to ice surfaces.
-
(2008) Biochimica et Biophysica Acta - Molecular Cell Research. 1783, 4, p. 557-566 Abstract
The Erv flavoenzymes contain a compact module that catalyzes the pairing of cysteine thiols into disulfide bonds. High-resolution structures of plant, animal, and fungal Erv enzymes that function in different contexts and intracellular compartments have been determined. Structural features can be correlated with biochemical properties, revealing how core sulfhydryl oxidase activity has been tailored to various functional niches. The introduction of disulfides into cysteine-containing substrates by Erv sulfhydryl oxidases is compared with the mechanisms used by NADPH-driven disulfide reductases and thioredoxin-like oxidoreductases to reduce and transfer disulfides, respectively.
-
(2008) Biochemistry. 47, 17, p. 4955-4963 Abstract
The flavoprotein quiescin-sulfhydryl oxidase (QSOX) rapidly inserts disulfide bonds into unfolded, reduced proteins with the concomitant reduction of oxygen to hydrogen peroxide. This study reports the first heterologous expression and enzymological characterization of a human QSOX1 isoform. Like QSOX isolated from avian egg white, recombinant HsQSOX1 is highly active toward reduced ribonuclease A (RNase) and dithiothreitol but shows a > 100-fold lower kcat/Km for reduced glutathione. Previous studies on avian QSOX led to a model in which reducing equivalents were proposed to relay through the enzyme from the first thioredoxin domain (C70-C73) to a distal disulfide (C509-C512), then across the dimer interface to the FAD-proximal disulfide (C449-C452), and finally to the FAD. The present work shows that, unlike the native avian enzyme, HsQSOX1 is monomeric. The recombinant expression system enabled construction of the first cysteine mutants for mechanistic dissection of this enzyme family. Activity assays with mutant HsQSOX1 indicated that the conserved distal C509-C512 disulfide is dispensable for the oxidation of reduced RNase or dithiothreitol. The four other cysteine residues chosen for mutagenesis, C70, C73, C449, and C452, are all crucial for efficient oxidation of reduced RNase. C452, of the proximal disulfide, is shown to be the charge-transfer donor to the flavin ring of QSOX, and its partner, C449, is expected to be the interchange thiol, forming a mixed disulfide with C70 in the thioredoxin domain. These data demonstrate that all the internal redox steps occur within the same polypeptide chain of mammalian QSOX and commence with a direct interaction between the reduced thioredoxin domain and the proximal disulfide of the Erv/ALR domain.
-
(2008) Journal of Agricultural and Food Chemistry. 56, 6, p. 2192-2198 Abstract
β-Casein is an intrinsically unstructured amphiphilic protein that self-assembles into micelles at neutral pH. This paper reports that β-casein self-organizes into micelles also under acidic conditions. The protein association behavior and micelle characteristics at pH 2.6, well below the p/, are presented. The pH was found to strongly affect the micelle shape and dimensions. Cryogenic transmission electron microscopy (cryo-TEM) experiments revealed disk-like micelles of 20-25 nm in length and ∼3.5 nm in height in acidic conditions. An aggregation number of 6 was determined by sedimentation equilibrium under these conditions. Isothermal titration calorimetry experiments verified the association below the p/ and allowed determination of the micellization enthalpy, the critical micellar concentration, and the micellization relative cooperativity (MR). Small-angle X-ray scattering results at concentrations below the critical micellization concentration (CMC) suggest that the monomeric protein is likely in a premolten globule state at low pH. Calculations of the protein charge at acidic and neutral pH reveal a similar high net charge but considerable differences in the charge distribution along the protein backbone. Overall the results show that β-casein is amphiphilic at low pH, but the distribution of charge along the protein chain creates packing constraints that affect the micelle organization, leading at concentrations above the CMC to the formation of disk micelles.
-
(2008) PROTEIN ENGINEERING DESIGN & SELECTION. 21, 2, p. 107-114 Abstract
We tested a disulfide-rich antifreeze protein as a potential scaffold for design or selection of proteins with the capability of binding periodically organized surfaces. The natural antifreeze protein is a β-helix with a strikingly regular two-dimensional grid of threonine side chains on its ice-binding face. Amino acid substitutions were made on this face to replace blocks of native threonines with other amino acids spanning the range of β-sheet propensities. The variants, displaying arrays of distinct functional groups, were studied by mass spectrometry, reversed-phase high performance liquid chromatography, thiol reactivity and circular dichroism and NMR spectroscopies to assess their structures and stabilities relative to wild type. The mutants are well expressed in bacteria, despite the potential for mis-folding inherent in these 84-residue proteins with 16 cysteines. We demonstrate that most of the mutants essentially retain the native fold. This disulfide bonded β-helical scaffold, thermally stable and remarkably tolerant of amino acid substitutions, is therefore useful for design and engineering of macromolecules with the potential to bind various targeted ordered material surfaces.
2007
-
(2007) Proceedings of the National Academy of Sciences of the United States of America. 104, 32, p. 13022-13027 Abstract
The interaction of membrane-embedded voltage-activated potassium channels (Kv) with intracellular scaffold proteins, such as the postsynaptic density 95 (PSD-95) protein, is mediated by the channel C-terminal segment. This interaction underlies Kv channel clustering at unique membrane sites and is important for the proper assembly and functioning of the synapse. In the current study, we address the molecular mechanism underlying Kv/PSD-95 interaction. We provide experimental evidence, based on hydrodynamic and spectroscopic analyses, indicating that the isolated C-terminal segment of the archetypical Shaker Kv channel (ShB-C) is a random coil, suggesting that ShB-C belongs to the recently defined class of intrinsically disordered proteins. We show that isolated ShB-C is still able to bind its scaffold protein partner and support protein clustering in vivo, indicating that unfoldedness is compatible with ShB-C activity. Pulldown experiments involving C-terminal chains differing in flexibility or length further demonstrate that intrinsic disorder in the C-terminal segment of the Shaker channel modulates its interaction with the PSD-95 protein. Our results thus suggest that the C-terminal domain of the Shaker Kv channel behaves as an entropic chain and support a "fishing rod" molecular mechanism for Kv channel binding to scaffold proteins. The importance of intrinsically disordered protein segments to the complex processes of synapse assembly, maintenance, and function is discussed.
-
(2007) Cell. 129, 2, p. 333-344 Abstract
Introduction of disulfide bonds into proteins entering the secretory pathway is catalyzed by Ero1p, which generates disulfide bonds de novo, and Pdi1p, which transfers disulfides to substrate proteins. A sufficiently oxidizing environment must be maintained in the endoplasmic reticulum (ER) to allow for disulfide formation, but a pool of reduced thiols is needed for isomerization of incorrectly paired disulfides. We have found that hyperoxidation of the ER is prevented by attenuation of Ero1p activity through noncatalytic cysteine pairs. Deregulated Ero1p mutants lacking certain cysteines show increased enzyme activity, a decreased lag phase in kinetic assays, and growth defects in vivo. We hypothesize that noncatalytic cysteine pairs in Ero1p sense the level of potential substrates in the ER and correspondingly modulate Ero1p activity as part of a homeostatic regulatory system governing the thiol-disulfide balance in the ER.
-
(2007) Journal of Biological Chemistry. 282, 6, p. 3458-3464 Abstract
Agrobacterium tumefaciens infects plant cells by the transfer of DNA. A key factor in this process is the bacterial virulence protein VirE2, which associates stoichiometrically with the transported single-stranded (ss)DNA molecule (T-strand). As observed in vitro by transmission electron microscopy, VirE2-ssDNA readily forms an extended helical complex with a structure well suited to the tasks of DNA protection and nuclear import. Here we have elucidated the role of the specific molecular chaperone VirE1 in regulating VireE2-VirE2 and VirE2-ssDNA interactions. VirE2 alone formed functional filamentous aggregates capable of ssDNA binding. In contrast, co-expression with VirE1 yielded monodisperse VirE1-VirE2 complexes. Cooperative binding of VirE2 to ssDNA released VirE1, resulting in a controlled formation mechanism for the helical complex that is further promoted by macromolecular crowding. Based on this in vitro evidence, we suggest that the constrained volume of the VirB channel provides a natural site for the exchange of VirE2 binding from VirE1 to the T-strand.
[All authors] -
(2007) Proceedings of the National Academy of Sciences of the United States of America. 104, 2, p. 462-466 Abstract
Intramembrane proteases catalyze peptide bond cleavage of integral membrane protein substrates. This activity is crucial for many biological and pathological processes. Rhomboids are evolutionarily widespread intramembrane serine proteases. Here, we present the 2.3-Å-resolution crystal structure of a rhomboid from Escherichia coli. The enzyme has six transmembrane helices, five of which surround a short TM4, which starts deep within the membrane at the catalytic serine residue. Thus, the catalytic serine is in an externally exposed cavity, which provides a hydrophilic environment for proteolysis. Our results reveal a mechanism to enable water-dependent catalysis at the depth of the hydrophobic milieu of the membrane and suggest how substrates gain access to the sequestered rhomboid active site.
2006
-
(2006) Journal of Molecular Biology. 362, 1, p. 89-101 Abstract
The ERV/ALR sulfhydryl oxidase domain is a versatile module adapted for catalysis of disulfide bond formation in various organelles and biological settings. Its four-helix bundle structure juxtaposes a Cys-X-X-Cys dithiol/disulfide motif with a bound flavin adenine dinucleotide (FAD) cofactor, enabling transfer of electrons from thiol substrates to non-thiol electron acceptors. ERV/ALR family members contain an additional di-cysteine motif outside the four-helix-bundle core. Although the location and context of this "shuttle" disulfide differs among family members, it is proposed to perform the same basic function of mediating electron transfer from substrate to the enzyme active site. We have determined by X-ray crystallography the structure of AtErv1, an ERV/ALR enzyme that contains a Cys-X4-Cys shuttle disulfide and oxidizes thioredoxin in vitro, and compared it to ScErv2, which has a Cys-X-Cys shuttle and does not oxidize thioredoxin at an appreciable rate. The AtErv1 shuttle disulfide is in a region of the structure that is disordered and thus apparently mobile and exposed. This feature may facilitate access of protein substrates to the shuttle disulfide. To test whether the shuttle disulfide region is modular and can confer on other enzymes oxidase activity toward new substrates, we generated chimeric enzyme variants combining shuttle disulfide and core elements from AtErv1 and ScErv2 and monitored oxidation of thioredoxin by the chimeras. We found that the AtErv1 shuttle disulfide region could indeed confer thioredoxin oxidase activity on the ScErv2 core. Remarkably, various chimeras containing the ScErv2 Cys-X-Cys shuttle disulfide were found to function efficiently as well. Since neither the ScErv2 core nor the Cys-X-Cys motif is therefore incapable of participating in oxidation of thioredoxin, we conclude that wild-type ScErv2 has evolved to repress activity on substrates of this type, perhaps in favor of a different, as yet unknown, substrate.
-
(2006) Protein Expression and Purification. 48, 2, p. 243-252 Abstract
The Tenebrio molitor thermal hysteresis protein has a cysteine content of 19%. This 84-residue protein folds as a compact β-helix, with eight disulfide bonds buried in its core. Exposed on one face of the protein is an array of threonine residues, which constitutes the ice-binding face. Previous protocols for expression of this protein in recombinant expression systems resulted in inclusion bodies or soluble but largely inactive material. A long and laborious refolding procedure was performed to increase the fraction of active protein and isolate it from inactive fractions. We present a new protocol for production of fully folded and active T. molitor thermal hysteresis protein in bacteria, without the need for in vitro refolding. The protein coding sequence was fused to those of various carrier proteins and expressed at low temperature in a bacterial strain specially suited for production of disulfide-bonded proteins. The product, after a simple and robust purification procedure, was analyzed spectroscopically and functionally and was found to compare favorably to previously published data on refolded protein and protein obtained from its native source.
-
(2006) Proceedings of the National Academy of Sciences of the United States of America. 103, 2, p. 299-304 Abstract
Ero1p is a key enzyme in the disulfide bond formation pathway in eukaryotic cells in both aerobic and anaerobic environments. It was previously demonstrated that Ero1p can transfer electrons from thiol substrates to molecular oxygen. However, the fate of electrons under anaerobic conditions and the final fate of electrons under aerobic conditions remained obscure. To address these fundamental issues in the Ero1p mechanism, we studied the transfer of electrons from recombinant yeast Ero1p to various electron acceptors. Under aerobic conditions, reduction of molecular oxygen by Ero1p yielded stoichiometric hydrogen peroxide. Remarkably, we found that reduced Ero1p can transfer electrons to a variety of small and macromolecular electron acceptors in addition to molecular oxygen. In particular, Ero1p can catalyze reduction of exogenous FAD in solution. Free FAD is not required for the catalysis of dithiol oxidation by Ero1p, but it is sufficient to drive disulfide bond formation under anaerobic conditions. These findings provide insight into mechanisms for regenerating oxidized Ero1p and maintaining disulfide bond formation under anaerobic conditions in the endoplasmic reticulum.
2005
-
(2005) Protein Science. 14, 6, p. 1630-1642 Abstract
Three different classes of thiol-oxidoreductases that facilitate the formation of protein disulfide bonds have been identified. They are the Ero1 and SOX/ALR family members in eukaryotic cells, and the DsbB family members in prokaryotic cells. These enzymes transfer oxidizing potential to the proteins PDI or DsbA, which are responsible for directly introducing disulfide bonds into substrate proteins during oxidative protein folding in eukaryotes and prokaryotes, respectively. A comparison of the recent X-ray crystal structure of Ero1 with the previously solved structure of the SOX/ALR family member Erv2 reveals that, despite a lack of primary sequence homology between Ero1 and Erv2, the core catalytic domains of these two proteins share a remarkable structural similarity. Our search of the DsbB protein sequence for features found in the Ero1 and Erv2 structures leads us to propose that, in a fascinating example of structural convergence, the catalytic core of this integral membrane protein may resemble the soluble catalytic domain of Ero1 and Erv2. Our analysis of DsbB also identified two new groups of DsbB proteins that, based on sequence homology, may also possess a catalytic core similar in structure to the catalytic domains of Ero1 and Erv2.
-
(2005) Proceedings of the National Academy of Sciences of the United States of America. 102, 13, p. 4729-4734 Abstract
We used cryo-electron tomography in conjunction with single-particle averaging techniques to study the structures of frozen-hydrated envelope glycoprotein (Env) complexes on intact Moloney murine leukemia retrovirus particles. Cryo-electron tomography allows 3D imaging of viruses in toto at a resolution sufficient to locate individual macromolecules, and local averaging of abundant complexes substantially improves the resolution. The averaging of repetitive features in electron tomograms is hampered by a low signal-to-noise ratio and anisotropic resolution, which results from the "missing- wedge" effect. We developed an iterative 3D averaging algorithm that compensates for this effect and used it to determine the trimeric structure of Env to a resolution of 2.7 nm, at which individual domains can be resolved. Strikingly, the 3D reconstruction is shaped like a tripod in which the trimer penetrates the membrane at three distinct locations ≃4.5 nm apart from one another. The Env reconstruction allows tentative docking of the x-ray crystal structure of the receptor-binding domain. This study thus provides 3D structural information regarding the prefusion conformation of an intact unstained retrovirus surface protein.
2004
-
(2004) Cell. 117, 5, p. 601-610 Abstract
The flavoenzyme Ero1p produces disulfide bonds for oxidative protein folding in the endoplasmic reticulum. Disulfides generated de novo within Ero1p are transferred to protein disulfide isomerase and then to substrate proteins by dithiol-disulfide exchange reactions. Despite this key role of Ero1p, little is known about the mechanism by which this enzyme catalyzes thiol oxidation. Here, we present the X-ray crystallographic structure of Ero1p, which reveals the molecular details of the catalytic center, the role of a CXXCXXC motif, and the spatial relationship between functionally significant cysteines and the bound cofactor. Remarkably, the Ero1p active site closely resembles that of the versatile thiol oxidase module of Erv2p, a protein with no sequence homology to Ero1p. Furthermore, both Ero1p and Erv2p display essential dicysteine motifs on mobile polypeptide segments, suggesting that shuttling electrons to a rigid active site using a flexible strand is a fundamental feature of disulfide-generating flavoenzymes.
2003
-
(2003) Journal of Virology. 77, 4, p. 2717-2729 Abstract
Infection of T lymphocytes by the cytopathic retrovirus feline leukemia virus subgroup T (FeLV-T) requires FeLIX, a cellular coreceptor that is encoded by an endogenous provirus and closely resembles the receptor-binding domain (RBD) of feline leukemia virus subgroup B (FeLV-B). We determined the structure of FeLV-B RBD, which has FeLIX activity, to a 2.5-Å resolution by X-ray crystallography. The structure of the receptor-specific subdomain of this glycoprotein differs dramatically from that of Friend murine leukemia virus (Fr-MLV), which binds a different cell surface receptor. Remarkably, we find that Fr-MLV RBD also activates FeLV-T infection of cells expressing the Fr-MLV receptor and that FeLV-B RBD is a competitive inhibitor of infection under these conditions. These studies suggest that FeLV-T infection relies on the following property of mammalian leukemia virus RBDs: the ability to couple interaction with one of a variety of receptors to the activation of a conserved membrane fusion mechanism. A comparison of the FeLV-B and Fr-MLV RBD structures illustrates how receptor-specific regions are linked to conserved elements critical for postbinding events in virus entry.
-
2002
-
(2002) Nature Structural Biology. 9, 1, p. 61-67 Abstract
Erv2p is an FAD-dependent sulfhydryl oxidase that can promote disulfide bond formation during protein biosynthesis in the yeast endoplasmic reticulum. The structure of Erv2p, determined by X-ray crystallography to 1.5 Å resolution, reveals a helix-rich dimer with no global resemblance to other known FAD-binding proteins or thiol oxidoreductases. Two pairs of cysteine residues are required for Erv2p activity. The first (Cys-Gly-Glu-Cys) is adjacent to the isoalloxazine ring of the FAD. The second (Cys-Gly-Cys) is part of a flexible C-terminal segment that can swing into the vicinity of the first cysteine pair in the opposite subunit of the dimer and may shuttle electrons between substrate protein dithiols and the FAD-proximal disulfide.
2000
-
(2000) Journal of Biological Chemistry. 275, 33, p. 25445-25450 Abstract
The GATE-16 protein participates in intra-Golgi transport and can associate with the N-ethylmaleimide-sensitive fusion protein and with Golgi SNAREs. The yeast ortholog of GATE-16 is the autophagocytosis factor Aut7p. GATE-16 is also closely related to the GABA receptor-associated protein (GABARAP), which has been proposed to cluster neurotransmitter receptors by mediating interaction with the cytoskeleton, and to the light chain-3 subunit of the neuronal microtubule-associated protein complex. Here, we present the crystal structure of GATE-16 refined to 1.8 Å resolution. GATE-16 contains a ubiquitin fold decorated by two additional N-terminal helices. Proteins with strong structural similarity but no detectable sequence homology to GATE-16 include Ras effectors that mediate diverse downstream functions, but each interacts with Ras by forming pseudo-continuous β-sheets. The GATE-16 surface suggests that it binds its targets in a similar manner. Moreover, a second potential protein-protein interaction site on GATE-16 may explain the adapter activity observed for members of the GATE-16 family.
1999
-
(1999) Proceedings of the National Academy of Sciences of the United States of America. 96, 26, p. 14759-14764 Abstract
Yeast Sec18p and its mammalian orthologue N-ethylmaleimide-sensitive fusion protein (NSF) are hexameric ATPases with a central role in vesicle trafficking. Aided by soluble adapter factors (SNAPs), Sec18p/NSF induces ATP-dependent disassembly of a complex of integral membrane proteins from the vesicle and target membranes (SNAP receptors). During the ATP hydrolysis cycle, the Sec18p/NSF homohexamer undergoes a large-scale conformational change involving repositioning of the most N terminal of the three domains of each protomer, a domain that is required for SNAP-mediated interaction with SNAP receptors. Whether an internal conformational change in the N-terminal domains accompanies their reorientation with respect to the rest of the hexamer remains to be addressed. We have determined the structure of the N- terminal domain from Sec18p by x-ray crystallography. The Sec18p N-terminal domain consists of two β-sheet-rich subdomains connected by a short linker. A conserved basic cleft opposite the linker may constitute a SNAP-binding site. Despite structural variability in the linker region and in an adjacent loop, all three independent molecules in the crystal asymmetric unit have the identical subdomain interface, supporting the notion that this interface is a preferred packing arrangement. However, the linker flexibility allows for the possibility that other subdomain orientations may be sampled.
-
(1999) Structure. 7, 6, p. 691-698 Abstract
Background: The hexameric helicase DnaB unwinds the DNA duplex at the Escherichia coli chromosome replication fork. Although the mechanism by which DnaB both couples ATP hydrolysis to translocation along DNA and denatures the duplex is unknown, a change in the quaternary structure of the protein involving dimerization of the N-terminal domain has been observed and may occur during the enzymatic cycle. This N-terminal domain is required both for interaction with other proteins in the primosome and for DnaB helicase activity. Knowledge of the structure of this domain may contribute to an understanding of its role in DnaB function. Results: We have determined the structure of the N-terminal domain of DnaB crystallographically. The structure is globular, highly helical and lacks a close structural relative in the database of known protein folds. Conserved residues and sites of dominant-negative mutations have structurally significant roles. Each asymmetric unit in the crystal contains two independent and identical copies of a dimer of the DnaB N-terminal domain. Conclusions: The large-scale domain or subunit reorientation that is seen in DnaB by electron microscopy might result from the formation of a true twofold symmetric dimer of N-terminal domains, while maintaining a head-to-tail arrangement of C-terminal domains. The N-terminal domain of DnaB is the first region of a hexameric DNA replicative helicase to be visualized at high resolution. Comparison of this structure to the analogous region of the Rho RNA/DNA helicase indicates that the N-terminal domains of these hexameric helicases are structurally dissimilar.
-
(1999) Molecular Cell. 3, 4, p. 487-493 Abstract
The E. coli Rho protein disengages newly transcribed RNA from its DNA template, helping terminate certain transcripts. We have determined the X- ray crystal structure of the RNA-binding domain of Rho complexed to an RNA ligand. Filters that screen both ligand size and chemical functionality line the primary nucleic acid-binding site, imparting sequence specificity to a generic single-stranded nucleic acid-binding fold and explaining the preference of Rho for cytosine-rich RNA. The crystal packing reveals two Rho domain protomers bound to a single RNA with a single base spacer, suggesting that the strong RNA-binding sites of Rho may arise from pairing of RNA- binding modules. Dimerization of symmetric subunits on an asymmetric ligand is developed as a model for allosteric control in the action of the intact Rho hexamer.
-
(1999) Nature Structural and Molecular Biology. 6, 4, p. 322-326 Abstract
Type II DNA topoisomerases mediate the passage of one DNA duplex through a transient break in another, an event essential for chromosome segregation and cell viability. The active sites of the type II topoisomerase dimer associate covalently with the DNA break-points and must separate by at least the width of the second DNA duplex to accommodate transport. A new structure of the Saccharomyces cerevisiae topoisomerase II DNA-binding and cleavage core suggests that in addition to conformational changes in the DNA-opening platform, a dramatic reorganization of accessory domains may occur during catalysis. These conformational differences have implications for both the DNA-breking and duplex-transport events in the topo II reaction mechanism, suggest a mechanism by which two distinct drug-resistance loci interact, and illustrate the scope of structural changes in the cycling of molecular machines.
1998
-
(1998) Proceedings of the National Academy of Sciences of the United States of America. 95, 14, p. 7876-7881 Abstract
Type IA and type II DNA topoisomerases are distinguished by their ability to cleave one or two strands, respectively, of a DNA duplex. Both types have been proposed to use an 'enzyme-bridging' mechanism, in which a break is formed in a DNA strand and a gap is opened between the broken pieces to allow passage of a second DNA strand or duplex segment. Although the type IA and type II topoisomerase structures appear overall quite different from one another, unexpected similarities between several structural elements suggest that members of the two subfamilies may use comparable mechanisms to bind and cleave DNA.
1997
-
(1997) Science. 277, 5332, p. 1662-1666 Abstract
An essential step in retrovirus infection is the binding of the virus to its receptor on a target cell. The structure of the receptor-binding domain of the envelope glycoprotein from Friend murine leukemia virus was determined to 2.0 angstrom resolution by x-ray crystallography. The core of the domain is an antiparallel β sandwich, with two interstrand loops forming a helical subdomain atop the sandwich. The residues in the helical region, but not in the B sandwich, are highly variable among mammalian C-type retroviruses with distinct tropisms, indicating that the helical subdomain determines the receptor specificity of the virus.