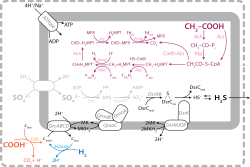
The chemical and isotopic composition of sedimentary rocks encodes information on fundamental events and processes in Earth history. Examples include the emergence and diversification of life, oxygenation of the atmosphere, and global-scale glaciations.
To extract this information, we must understand (i) the processes responsible for generating geochemical and isotopic signals, (ii) the processes that lead to their preservation in the rock record, and (iii) those that alter the original signals to yield the observed chemical and isotopic composition of sedimentary rocks.
My group decodes these signals by combining geochemical and geological observations, laboratory experiments, theory, and numerical models of variable complexity. The problems we study span timescales of seconds to billions of years, and spatial scales of a single microbe to an entire planet.
Contact me for research opportunities related to these topics.