Asher Lab


Research
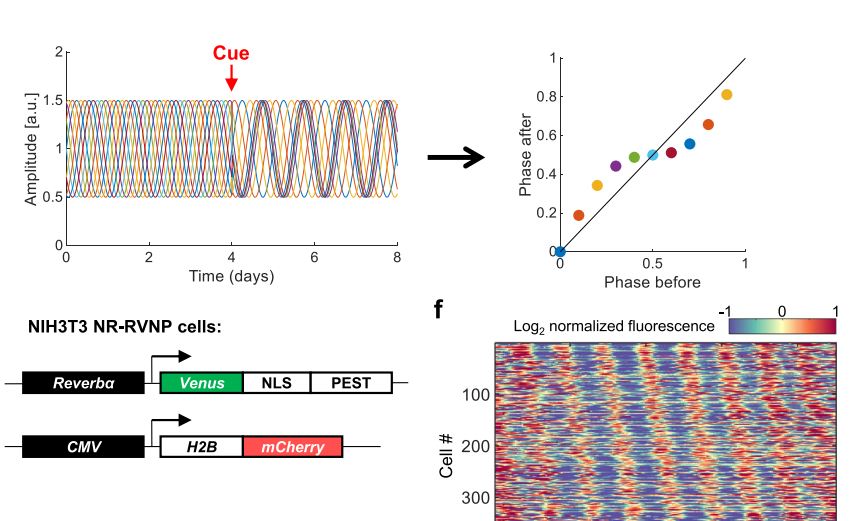
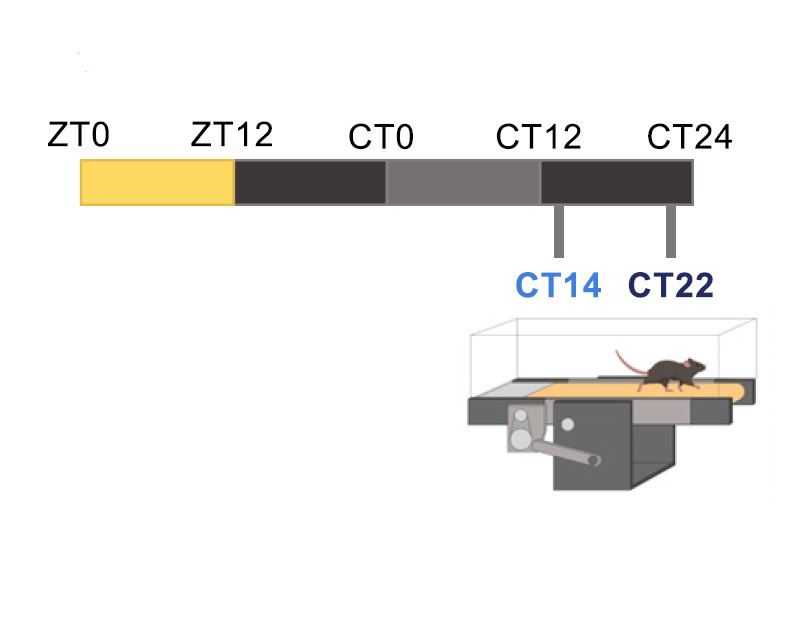
Circadian rhythms, 24-hour cycles in behavior, physiology, and metabolism, are a fundamental feature of life on Earth. From cyanobacteria to humans, all light-sensitive organisms have evolved an internal clock that helps them anticipate daily changes in their environment.
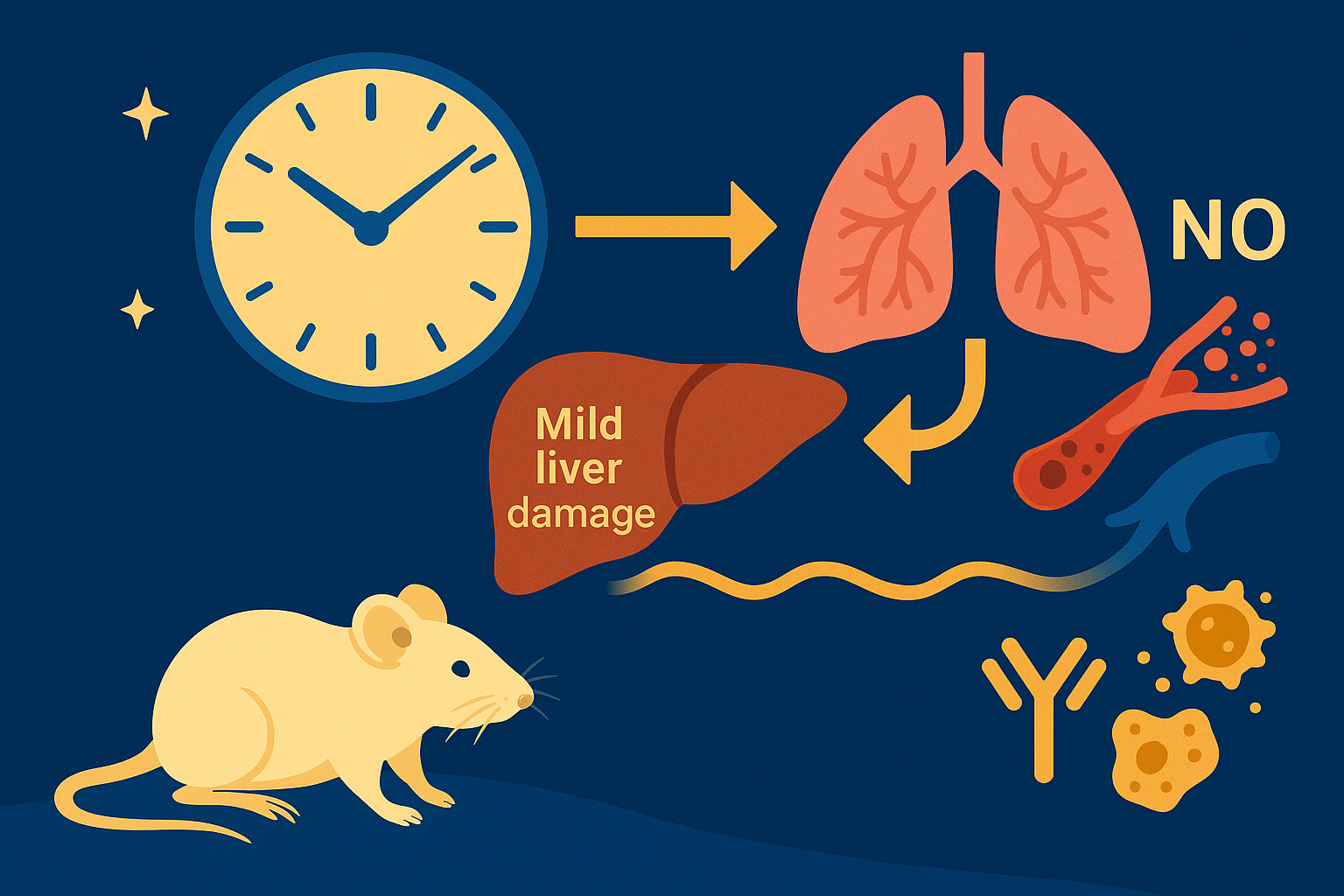
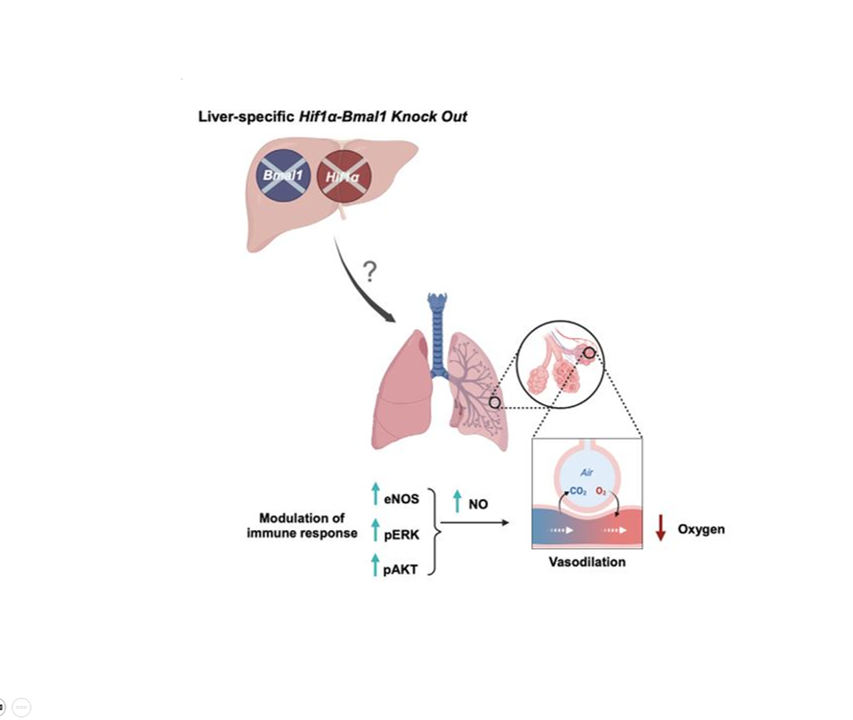
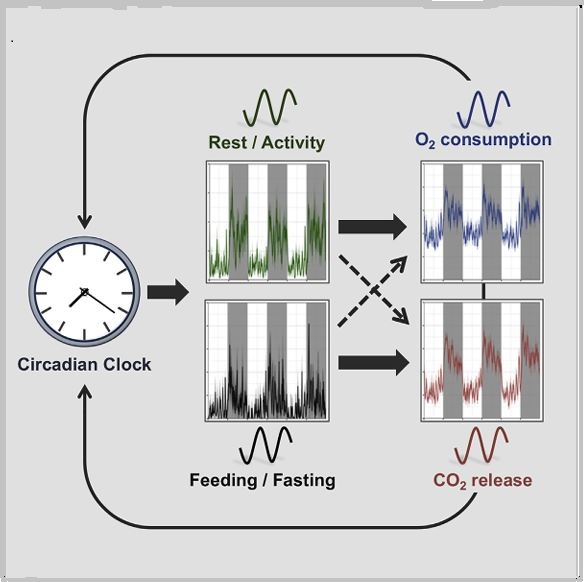
In mammals, this system is coordinated by a central clock in the brain, which keeps time with the day-night cycle. But that's only part of the story as most cells in our body harbor their own “peripheral” clock. These peripheral clocks are synchronized with each other through various time-signals (such as food intake, oxygen levels, and temperature). Dis-synchrony between them and the environment is associated with a wide variety of pathologies, from sleep apnea to metabolic syndrome and cancer.
Recent breakthroughs have uncovered an intricate interplay between circadian rhythms and metabolism. Our internal clocks not only influence our metabolism throughout the day but are also shaped by our metabolic state. Disruption of circadian rhythms is widely implicated in various metabolic diseases such as obesity, diabetes, and metabolic syndrome.
In our lab, we explore the intricate relationship between circadian biology and metabolism from the molecular through cellular and whole organism levels. We are interested in understanding how this interplay contributes to health and disease. Through our research, we hope to uncover new insights that will inform treatments and lifestyle strategies for improving metabolic health.