-
News
 Sunday, December 19, 2021
Sunday, December 19, 2021Farewell to Drs. Tong Bian and Damian Myśliwiec
Date:Sunday, December 19, 2021 -
Publications
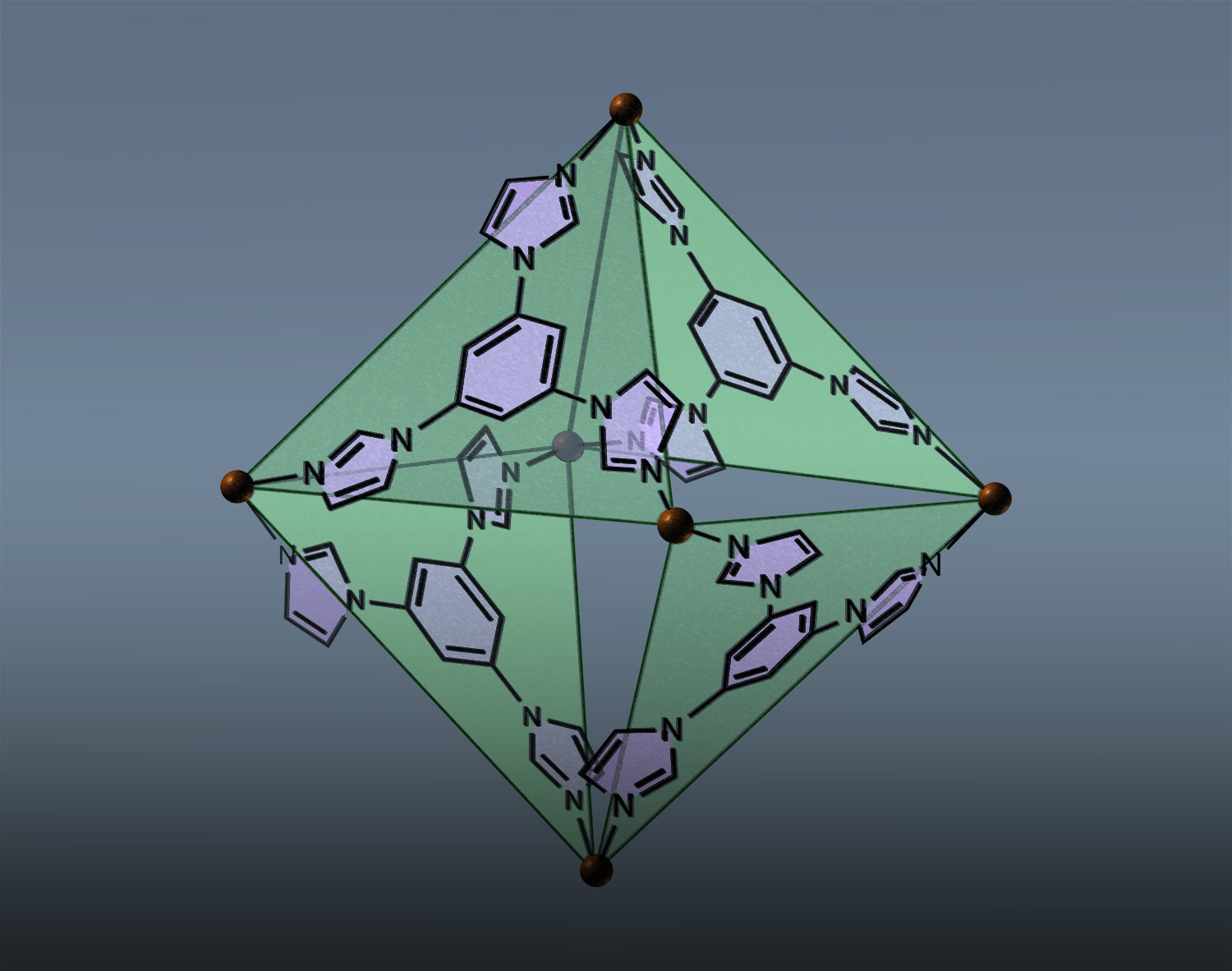
Guest encapsulation alters the thermodynamic landscape of a coordination host
-
Publications
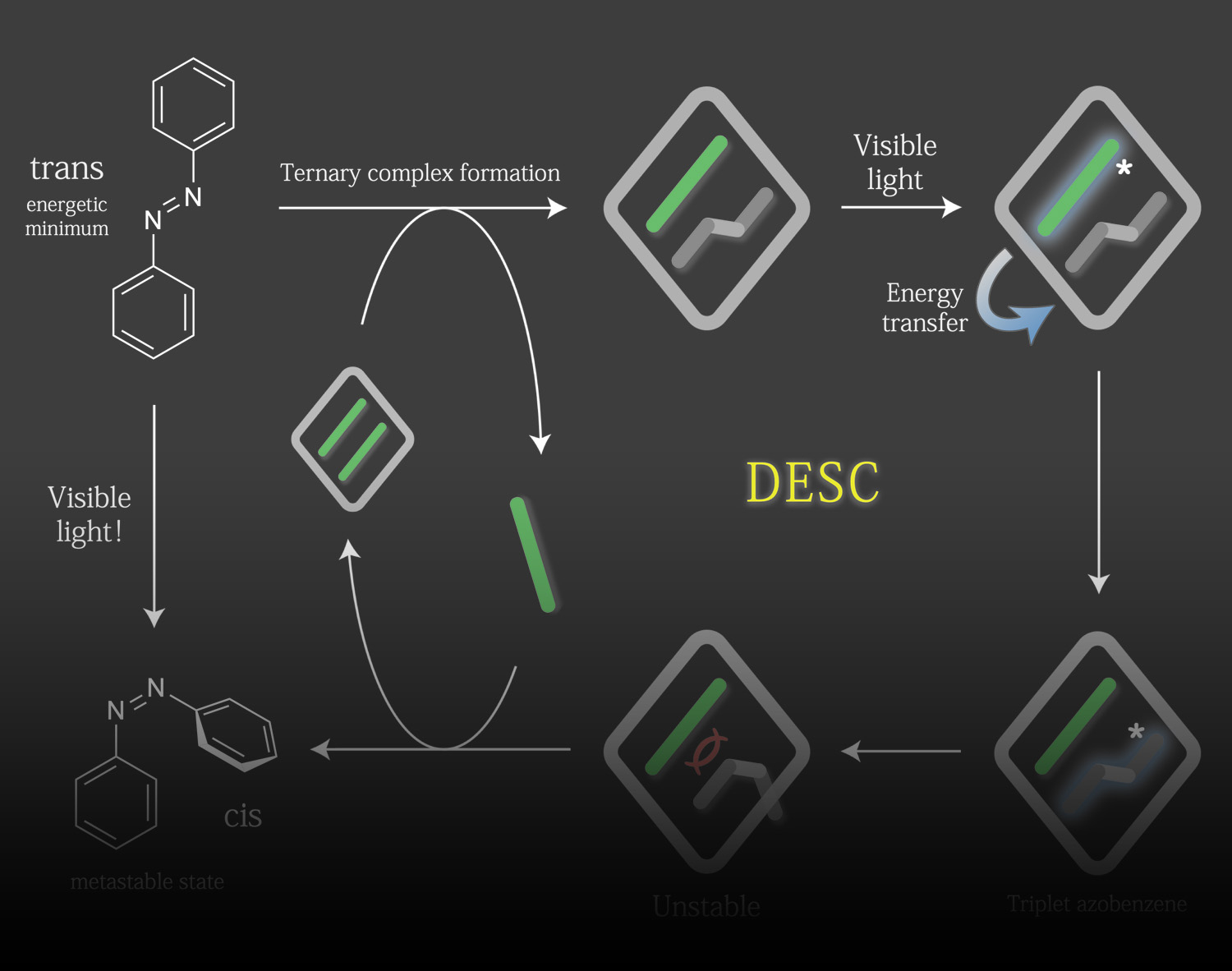
Disequilibrating azobenzenes by visible-light sensitization under confinement
-
Publications
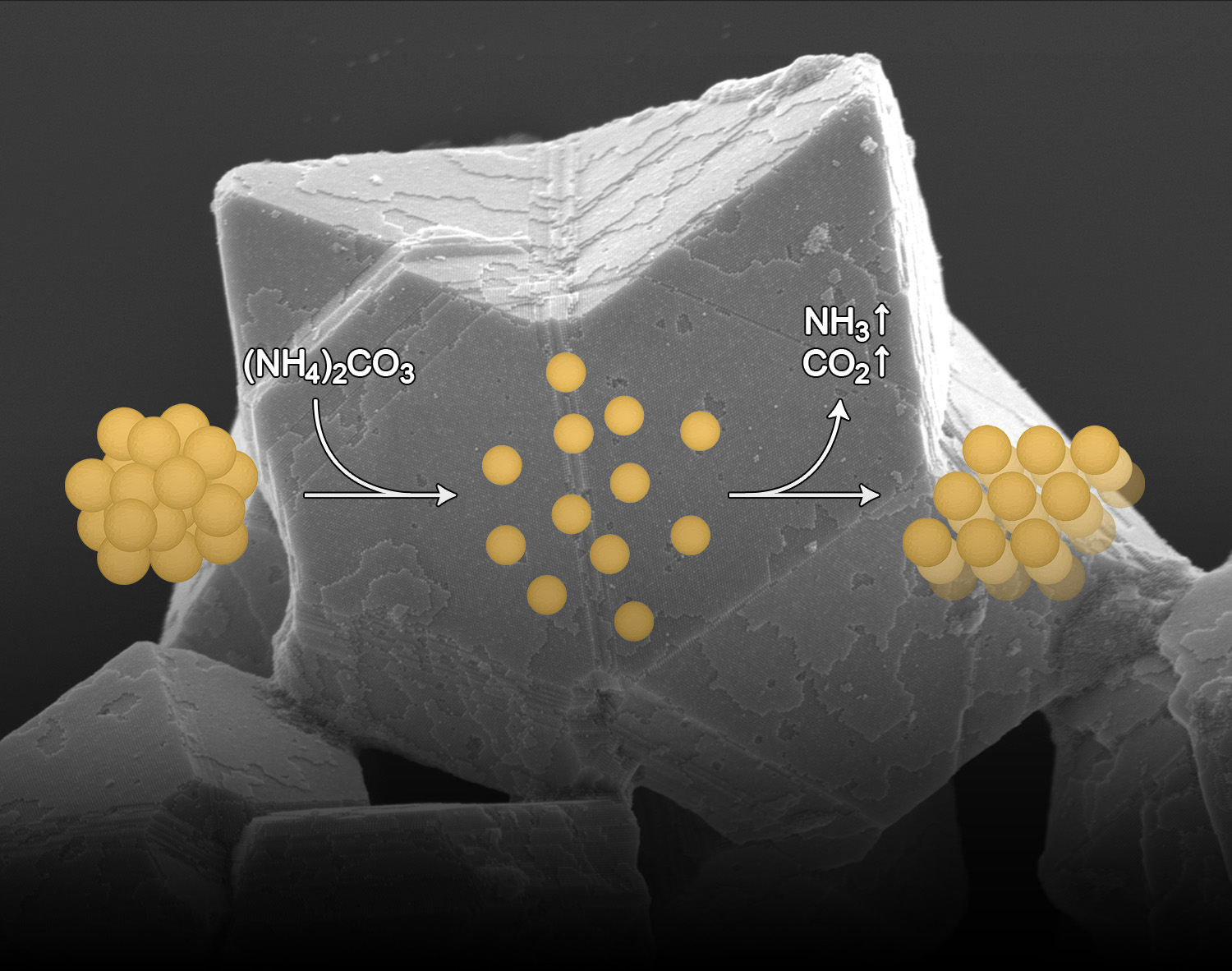
Photocleavable anionic glues for light-responsive nanoparticle aggregates