
Professor Edna Mozes
Professor of Immunology
E-mail: edna.mozes@weizmann.ac.il
Phone: +972-8-934-3646
Fax: +972-8-934-4173
Location: Wolfson Bldg., Room 526
|
Professor Edna Mozes
E-mail: edna.mozes@weizmann.ac.il
|
Research Interests:
Specific Immunomodulation of Autoimmune Diseases
Our main objectives are to shed light on the mechanisms underlying the amelioration of two autoimmune diseases, namely, systemic lupus erythematosus (SLE) and myasthenia gravis (MG), by peptides that we designed as novel means for the specific treatment of these diseases.
Myasthenia gravis (MG) (in collaboration with Michael Sela)
Myasthenia gravis (MG) and its experimental model, experimental autoimmune MG (EAMG), are immune disorders that are characterized by circulating antibodies and lymphocyte autoreactivity to the nicotinic acetylcholine receptor (AChR), leading to a reduced number of AChR molecules at the postsynaptic end plates. Two myasthenogenic peptides, p195-212 and p259-271, that are sequences of the human AChR -subunit were shown to induce MG-associated immune responses in mice. A dual altered peptide ligand (APL) composed of the two single amino acids substituted analogs of the two myasthenogenic peptides inhibited in vitro and in vivo those responses. The suppressive properties of the dual APL were also shown following immunization with the whole Torpedo AChR and it down-regulated the clinical manifestations of an established EAMG induced either in mice or in rats by the Torpedo AChR. The in vitro and in vivo inhibiting effects of the dual APL were associated with a significant down-regulation of the pathogenic cytokine IFN-γ and with an up-regulation of TGF-β and IL-10. Furthermore, the suppressive effect of the dual APL could be transferred to mice, immunized with either the myasthenogenic peptides or with the AChR, by their inoculation with immunocytes of dual APL treated mice.
The dual APL acts by up-regulating CD4+ CD25+ cells that express characteristic regulatory markers such as CD45RBlow, Foxp3, CTLA-4, intracellular and membrane-bound TGF-β. Further studies demonstrated that CD8 regulatory cells are also involved in the suppressive function of the dual APL. Investigation of the signaling events induced by the dual APL, showed an association between the levels of TGF-β and JNK activity. JNK protein is known to activate the transcription of FasL gene which initiates activation induced cell death. As expected, treatment with the dual APL increased the apoptotic rate as measured by the up-regulation of the key markers Fas, FasL, caspases 3 and 8 and the down-regulation of the anti-apoptotic markers cFLIP and Bcl-2. (Figure 1).
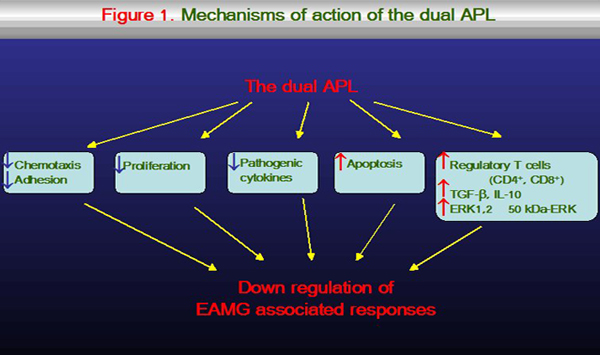
Furthermore, the dual APL was shown to up-regulate the phosphorylation of ERK1,2 in CD4+ CD25+ cells, whereas it decreased the phosphorylation of ERK1,2 in CD4+ CD25- cells. Inhibition of ERK1,2 in the dual APL induced CD4+ CD25+ cells, abrogated their ability to suppress MG-associated responses. Moreover, inhibition of ERK1,2 in the dual APL induced CD4+ CD25+ cells, down-regulated Foxp3 expression, and CD4+ CD25+ regulatory cell development, suggesting that the active suppression exerted by the dual APL depends on ERK1,2 activity in CD4+ CD25+ cells. In addition, the dual APL up-regulated a novel 50 kDa ERK in the induced CD4+ CD25+ cell population (Figure 1). Thus, the ability of the dual APL to interfere with signaling associated events results in its significant therapeutic potential.Systemic lupus erythematosus (SLE)
SLE is an autoimmune disease characterized by an increased production of autoantibodies and systemic clinical manifestations. For a specific treatment of SLE we have designed and synthesized a peptide (hCDR1) based on the complementarity determining region (CDR) 1 of an anti-DNA autoantibody and tested its effects on several mice models for lupus:
Treatment with hCDR1 ameliorated the serological and clinical manifestation of the established lupus in these models. Further, hCDR1 down-regulated disease manifestations associated with neurological lupus. The beneficial effects of hCDR1 were associated with a decreased secretion and expression of the pathogenic cytokines, IFN-γ, IL-10, IL-1β and TNF-α and with an up-regulation of the immunosuppressive cytokine, TGF-β.
- a) the spontaneous model of (NZBxNZW)F1 mice,
- b) induced experimental SLE in BALB/c mice, and
- c) a �humanized� model of severe combined immunodeficient (SCID) mice engrafted with PBL of SLE patients.
Treatment with hCDR1 up-regulated the CD4+ CD25+CD45RBlow cells with regulatory characteristics, including the expression of Foxp3, CTLA-4 and TGF-β (Figure 2).
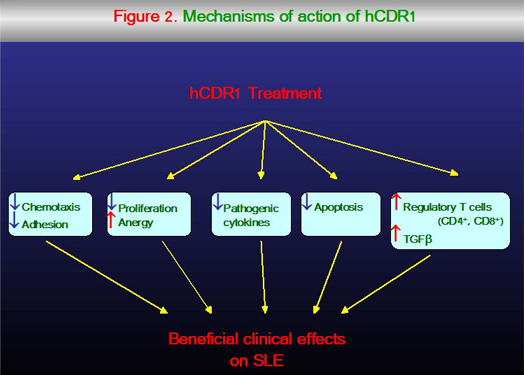
Adoptive transfer of hCDR1-treated splenocytes to old (NZBxNZW)F1 mice with a full blown disease resulted in a reduction of the anti-dsDNA autoantibodies and in the amelioration of the kidney disease in the recipient mice. Depletion of the CD25 expressing cells diminished significantly the therapeutic effects of hCDR1, whereas administration of the enriched CD4+ CD25+ cell population was beneficial to the diseased mice. The hCDR1-induced CD4CD25 expressing cells suppressed the activation of autoreactive CD4+ cells as indicated by the diminished expression of CD69 and Fas ligand on the latter, resulting in reduced rates of activation-induced apoptosis. The latter was also indicated by the reduced expression of caspases 3 and 8 and by the up-regulation of the anti apoptotic molecule Bcl-xL, which had tolerogenic effects on SLE. In addition, treatment with hCDR1 resulted in the diminished expression of the pro-apoptotic factor JNK kinase along the p21Ras/MAP kinase pathway. Treatment with hCDR1 up-regulated CD8+ regulatory cells as well. The latter cells were shown to be essential for the induction and the optimal suppressive function of CD4+ CD25+ cells following treatment with hCDR1 (Figure 2).The immunosuppressive cytokine, TGF-β, was shown to play a key role in the inhibition by hCDR1-induced CD4+ CD25+ cells. This cytokine was demonstrated to be secreted by CD4+ cells that were affected by hCDR1-induced immunoregulatory T cells. The up-regulated secretion of TGF-β as well as the down-regulation of activated autoreactive cells was associated with a decrease in the pathogenic cytokines (e.g. IFN-γ, IL-10). Treatment with hCDR1 down-regulated T cell adhesion and chemotaxis via the up-regulated TGF-β. The latter was associated with the down-regulation of ERK phosphorylation (known to participate in the signalling cascade that is involved in cell locomotion). Further, hCDR1 induced suppression of ERK-phosphorylation resulted in the diminished expression and function of a pair of key cell adhesion receptors, LFA-1 and CD44, which operate as accessory molecules in mediating APC-T-cell interactions. Finally, hCDR1 was shown to inhibit TCR signalling (ZAP-70 phosphorylation) and to up-regulate two negative regulators of TCR activation, namely Foxj1 and Foxo3a and two early growth response genes, namely Egr-2 and Egr-3 that are required for the induction of anergy (Figure 2). Thus, treatment with hCDR1 leads to a cascade of events that culminates in the down-regulation of SLE-associated autoreactive responses and in the clinical improvement of experimental SLE. The hCDR1 is therefore a potential candidate for a novel specific treatment of SLE patients.
Molly Dayan, Ph.D., Lab assistant
Heidy Zinger, B.A., Lab Aassistant
Ezra Vadai, Lab Aassistant
Hava Ben-David, Ph.D., Post-doctoral Fellow
Smadar Gertel, Ph.D., Post-doctoral Fellow
Reshmi Parameswaran, Ph.D., Post-doctoral Fellow
Uri Sela, M.D., Ph.D., Post-doctoral Fellow
Amir Sharabi, M.D., Ph.D., Post-doctoral Fellow
Prof. Zev Sthoeger, M.D., Visitng Scientist