


This review was write by Prof. Kurt Giles, while he was a post-doc in the Silman/Sussman lab in 1996-99.
History
Dale's momentous 1914 paper, in which he differentiated between the muscarine- and nicotine- like actions of choline esters on different tissues, proposed: "it seems not improbable that an esterase contributes to the removal of [acetylcholine] from the circulation". This hypothesis was based on observations of the inactivation of acetylcholine (ACh) injected into cats. However, it wasn't until 1926 that Loewi and Navratil, working on isolated frog's hearts, experimentally demonstrated its existence by inhibition with physostigmine (eserine), thus prolonging the effect of administered ACh. In 1932 Stedman et al. prepared a crude extract of an ACh-splitting enzyme from horse serum, which they called "cholinesterase."
Cholinesterases from different species were found to differ in their substrate specificity and susceptibility to inhibitors. This provoked numerous schemes for naming the various cholinesterases. They are now considered to constitute a family of enzymes which fall broadly into two types depending on their substrate preference (Silver, 1974). This division is not absolute and holds true more in mammalian than non-mammalian species. Those enzymes which preferentially hydrolyse acetyl esters such as ACh are called acetylcholinesterase (AChE) or acetylcholine acetylhydrolase (EC 3.1.1.7), and those which prefer other types of esters such as butyrylcholine are termed butyrylcholinesterase (BChE) or acylcholine acylhydrolase (EC 3.1.1.8). BChE is also known as pseudocholinesterase, non- specific cholinesterase, or simply cholinesterase. This last term has led to confusion, and in this thesis the term cholinesterases will refer to all choline ester hydrolysing enzymes, irrespective of their substrate specificity.
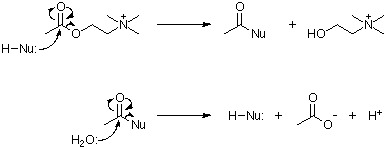
The main function of AChE is the rapid hydrolysis of the neurotransmitter ACh at cholinergic synapses. The hydrolysis reaction proceeds by nucleophilic attack of the carbonyl carbon, acylating the enzyme and liberating choline. This is followed by a rapid hydrolysis of the acylated enzyme yielding acetic acid, and the restoration of the esteratic site (Wilson et al., 1950).
{kind=link}
The function of BChE remains a puzzle, it has no known specific natural substrate, although it is capable of hydrolysing ACh. It has been suggested that BChE acts as a scavenging enzyme in the detoxification of natural compounds (Massoulié et al., 1993). Certain human individuals have a mutant BChE which lacks the ability to hydrolyse succinyl choline. In rare individuals the complete BChE gene is missing. Neither of these cases result in any apparent physiological consequence. There is however an important clinical implication; succinyl choline is commonly used during tracheal intubation in the administration of inhalation anaesthetics, and causes post operative apnoea in these people (McGuire et al., 1989).
Active site structure
The traditional view of the active site of AChE was considered to consist of two subsites; a negatively charged or 'anionic' site, to which the positively charged quaternary nitrogen moiety binds, and an esteratic site containing the actual catalytic residues, probably both an electrophilic and a nucleophilic group (Nachmansohn and Wilson, 1951). A second 'anionic' site, which became known as the 'peripheral anionic' site, around 14Å from the active site, was proposed on the basis of binding of bis quaternary compounds (Bergmann et al., 1950). O'Brien (1969) proposed his "heretical thought" that no true anionic site existed. He argued that the carbon analogue of ACh which lacks the charge of the quaternary nitrogen atom, and other uncharged molecules, are good substrates for the enzyme. He also noted that if coulombic forces were of major importance in substrate binding, then the ammonium ion should be a very good inhibitor, whereas it is one of the poorest.
{kind=link}
The nucleophile was assumed to be a serine residue, with a histidine residue enhancing its nucleophilicity (Cunningham, 1957). As other enzyme mechanisms became understood, AChE was classified as a serine hydrolase, and therefore assumed to contain a catalytic triad of Asp-His-Ser at the esteratic subsite.
AChE from the electric ray Torpedo californica has most recently been the centre of attention due to its high concentration in the electroplax. According to the agreement adopted at the Oholo Conference in 1992, residues are numbered from the first residue of the mature protein. All references to residue number are for AChE from T. californica unless otherwise stated, when the number of the homologous residue of T. californica AChE is given in italics, in parentheses (Massoulié et al., 1992a).
The position of the active site serine in T. californica AChE was established by irreversibly labelling it with [3H]isopropyl fluorophosphate, followed by tryptic digestion and analysis of the tryptic peptides (MacPhee-Quigley et al., 1985; Schumacher et al., 1986), localising it to Ser200. Mutagenesis studies identified the catalytic histidine (Gibney et al., 1990) as His440. The aspartic acid group of the catalytic triad was assumed to be Asp326.
Crystal structure
A great leap forward in the understanding of the catalytic mechanism, and mode of action of inhibitors, came in 1991 with the determination of the three dimensional structure of dimeric T. californica AChE (Sussman et al.). This enzyme was crystallised in 1988 (Sussman et al.), and its structure was determined to a resolution of 2.8Å (Sussman et al., 1991), which has more recently been refined to 2.2Å (J.L. Sussman, personal communication).
The structure determination uncovered a number of interesting findings. The catalytic triad seemed to contain a glutamate residue, rather than the usual aspartate. This was confirmed by site- directed mutagenesis on the closely related T. marmorata AChE; the mutations Glu327Gln and Glu327Asp led to inactive products (Duval et al., 1992). The relation of this triad to the rest of the protein approximates a mirror image of that seen in the serine proteases. The only other proteins known with a similar catalytic triad, are fungal lipases from Geotrichum candidum (Schrag et al., 1991) and Candida rugosa (Grochulski et al., 1993). Both these enzymes show a high overall similarity with the primary and tertiary structures of AChE. These proteins have recently been classified as members of a new protein fold; the α/β hydrolase fold (Ollis et al., 1992). α/β hydrolase fold proteins are considered to be related to the trypsin family of serine proteases, subtilisin and papain by convergent evolution.
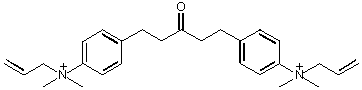
The active site was found to be located 20Å from the enzyme surface at the bottom of a narrow gorge, lined with 14 aromatic residues, which may be important in guiding the substrate to the active site (Ripoll et al., 1993). There was no discernible 'anionic' site, the quaternary nitrogen of choline binds chiefly through interactions with the pi electrons of the residue Trp84 (Sussman et al., 1991). The structures of AChE with the bound inhibitors; decamethonium, tetrahydroaminoacridine (Tacrine) and edrophonium (Harel et al., 1993), and 1,5-bis (4-allyldimethylammoniumphenyl) pentan-3-one dibromide (BW284c51; J.L. Sussman, personal communication), have been determined. These show that ligands binding at the peripheral site also do so by interaction with π electrons, in this instance with the residue Trp279.
Inhibition
Inhibition can be either reversible, by competitively blocking the substrate reaching the active site; or quasi-irreversible, by covalent reaction with the active site serine, inactivating the catalytic ability of the enzyme.
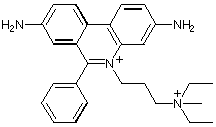
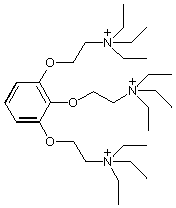
Competitive inhibition takes place by blocking substrate at the active site (Tacrine, edrophonium); non-competitive inhibition occurs by binding to the peripheral site (propidium, gallamine). The bis-quaternary ligands decamethonium and BW284c51 bind across both active and peripheral sites.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
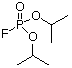
The longest known and most widely used inhibitor is the natural alkaloid physostigmine. This carbamoylates the active site serine residue, greatly slowing the acyl-enzyme hydrolysis reaction compared with the acetylated enzyme. Organophosphorus compounds such as diisopropyl fluorophosphate (DFP) are very potent inhibitors of AChE and are used as agricultural insecticides or as nerve gases in chemical warfare. These compounds react with the active site serine forming a very stable covalent phosphoryl-enzyme complex.
){kind=link}
{kind=link}
Another potent AChE inhibitor is fasciculin, a toxin from green mamba snake venom, its crystal structure has recently been determined (le Du et al., 1992). Computer modelling and mutation studies of fasciculin and AChE have localised the area of interaction to the aromatic residues near the rim of the gorge (Radic et al., 1994).
Genetics
Up to three genes for AChE have been found in invertebrates. Mutants of Caenorhabditis elegans lacking any two of the three are still viable, despite phenotypic differences (Arpagaus et al., 1994). However, classification as true AChE is sometimes difficult due to the enzyme's substrate specificity. Vertebrates all contain a single AChE gene, whose structure differs slightly with taxonomic group (Massoulié et al., 1992a). mammalian AChE gene (Taylor, 1992) is located at 7q22 in humans (Getman et al., 1992).
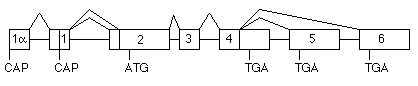{kind=link}
The gene is unusual in containing two transcription origins; the transcript including 1a has been found only in brain and is possibly a developmental form (P. Taylor, personal communication). At the 3' end of the coding region, exon 4 is either read through to give the R subunit, or alternatively spliced to exon 5 (H subunit) or exon 6 (T subunit). The R transcript has so far only been observed in mouse erythroid cells (Li et al., 1993). The mature catalytic subunit of AChE consists of a major common domain of about 535 residues, followed by variable C-terminal peptides due to this alternative splicing.
Despite genomic differences, the primary structure of many cholinesterases are highly homologous to enzymes as evolutionary divergent as some fungal lipases.
Quaternary Structure
Cholinesterases show a polymorphism of quaternary structures, of similar catalytic activity but differing in their hydrodynamic parameters and ionic or hydrophobic interactions. Historically the different polymorphic forms have been separated by sedimentation on density gradients, with or without detergent, which has led to a number of naming strategies. In this review molecular forms will be referred to using the recently agreed upon nomenclature (Massoulié et al., 1992a).
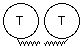
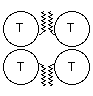
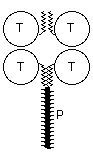
Catalytic subunits, which may vary in glycosylation, can oligomerise into dimers or tetramers, giving rise to the globular (G) forms: G1, G2 and G4. The globular forms can further be divided depending on their amphiphilicity, due to the possibility of cell membrane attachment by either a glycophosphatidylinositol (GPI) anchor or the hydrophobic P subunit. Attachment of a collagen-like tail (Q subunits) to one, two or three catalytic tetramers gives the A4, A8 and A12 asymmetric forms which bind to the basal lamina. Free cysteine residues form inter-subunit disulphide bonds, to covalently dimerise catalytic subunits, and to attach catalytic subunits to P and Q polypeptide anchors. Tetramers are formed by electrostatic and hydrophobic interaction between two disulphide bonded dimers.
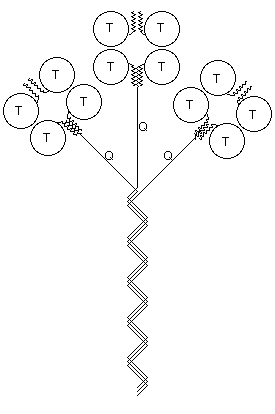{kind=link}
Membrane Attachment
In humans, H subunits are unique to cells of the haematopoietic sequence, they form amphiphilic dimers G2a type I (Massoulié et al., 1992b) and each subunit has its C-terminal residues trimmed, and a GPI membrane anchor attached. Human G2a type I AChE, unlike G2a type I from other sources, is not released by a bacterial phosphatidylinositol specific phospholipase C (PI-PLC). This resistance is due to the existence of an additional palmitoyl group on an inositol hydroxyl (Roberts et al., 1988b). Deacylation of human erythrocyte AChE by treatment with hydroxylamine rendered it susceptible to PI-PLC (Toutant et al., 1989). Although serum anchor-specific phospholipase D (PLD) cleaves the intact human erythrocyte AChE anchor, it does not release amphiphilic dimers from the membrane, and hydroxylamine pretreatment is again required (Toutant et al., 1989). The structure of this anchor has recently been determined. N.B. The assignment of the position of the palmitoyl group on the inositol ring is not known, hindrance of PI-PLC suggests that it may be next to the phosphate group.
{kind=link}
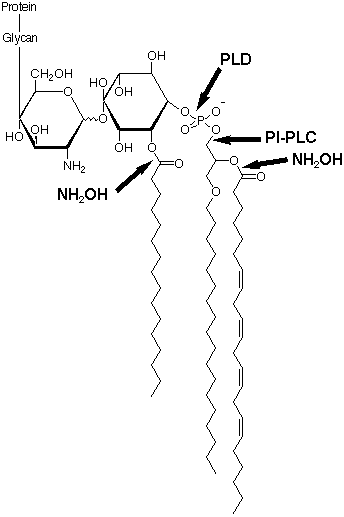{kind=link}
The T subunits form amphiphilic monomers G1a type II and dimers G2a type II which can associate with cellular membranes but are easily solubilised without detergents. Palmytoylation has been suggested as a possibility for membrane attachment (Massoulié et al., 1993) however a free cysteine residue, necessary for thioester bond formation, is only available in G1a type II monomers. These monomers aggregate on solubilisation unlike G2a type II dimers. Tetramers of the T subunits may either be the fully soluble non-amphiphilic G4na, or require detergent for solubilisation G4a). G1a type II, G2a type II, G4a and G4na are generally found in the CNS. Asymmetric forms, which also consist of T subunits, are found postsynaptically at neuromuscular junctions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The proposed association of the G1a type II and G2a type II structures with the cell membrane (Massoulié et al., 1993) is surprising in light of a Kyte and Doolittle hydropathicity plot of the forty C-terminal residues of the T subunit of human and bovine AChE, which show them to be largely hydrophilic. This region is highly conserved, being identical in human and bovine and showing a 75% identity with T. californica AChE.
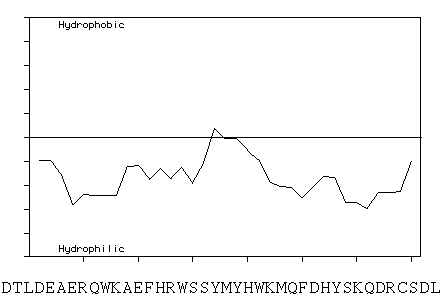{kind=link}
In mammalian brain nearly all AChE activity is due to G4a (Muller et al., 1985), membrane anchored with the hydrophobic P subunit. The P subunit was first identified in bovine brain as a 20kDa protein (Inestrosa et al., 1987). A 13kDa fragment, released upon treatment with proteinase K, was shown to be responsible for hydrophobic aggregation of G4a AChE (Fuentes et al., 1988). This subunit has also been demonstrated in rat brain (Boschetti et al., 1994), monkey brain (Liao et al., 1993) and human caudate nucleus (Gennari et al., 1987). A similar structural protein has been reported in the nematodes Steinernema carpocapsae (Arpagaus et al., 1992) and C. elegans.
Substrate Inhibition
The inhibition of AChE by excess substrate is one of the key features that distinguishes it from BChE. BChE exhibits the converse substrate activation, and both phenomena are likely to be related to the binding of substrate, and to the catalytic mechanism of the enzymes. It is not known whether substrate inhibition has a biological role, or is simply a consequence of the structure and mechanism of AChE.
Various explanations have been proposed to account for the different responses of AChE and BChE to excess substrate. Rosenberry (1975) suggested that the rate limiting step in catalysis was formation of an induced fit complex, and substrate inhibition was brought about by interference with this step. It has also been proposed that substrate inhibition is due to deacylation being retarded by binding of a second ACh molecule to the anionic subsite of the acyl enzyme (Krupka, 1963).
It has been suggested that AChE is allosterically regulated by the binding of ACh to the peripheral site through conformational changes at the active centre (Changeux, 1966). During development, the electrophoretic mobility and sedimentation coefficient of rat brain G4a AChE remain constant, but its kinetic parameters including substrate inhibition change (Inestrosa and Ruiz, 1985). Amphiphilic AChE from mosquito showed no substrate inhibition when freshly extracted, however upon conversion to its non-amphiphilic derivatives, with thiocyanate (a chaotropic anion), the substrate inhibition returned (Dary and Wedding, 1990). These results indicate that the environment may confer a slight conformational change in AChE, which results in substrate inhibition.
The importance of the peripheral site in substrate inhibition has been supported by Radic et al. (1991), based on studies of competition of substrate with the peripheral site ligand propidium. However studies on chicken AChE, which lacks the Tyr-70 and Trp-279 residues of the T. californica peripheral site, reveal substrate inhibition characteristics similar to those of T. californica AChE (Eichler et al., 1994).
The mutations carried out so far, which abolish substrate inhibition, can be divided into two groups; those at the active site, and those at the peripheral site. Mutations of recombinant human AChE (rhAChE) at the active site have concentrated on the glutamate residue next to the catalytic serine. Mutations Glu-202(199)Asp, Glu-202(199)Gln and Glu-202(199)Ala all abolish substrate inhibition (Shafferman et al., 1992), the same effect has also been demonstrated by mutation Glu- 199Gln in T. marmorata AChE (Gibney et al., 1990).
In rhAChE, mutants of an aspartate at the gorge entrance Asp-74(72)Asn, Asp-74(72)Gly and Asp-74(72)Lys all abolish substrate inhibition, possibly by inducing a conformational change. Mutation of a gorge entrance tryptophan residue Trp-286(279)Ala reduces substrate inhibition, probably by a similar mechanism (Shafferman et al., 1992). Mutations on mouse AChE have also been studied, Phe-297(290)Ile not only eliminates substrate inhibition, but confers elements of substrate activation characteristic of BChE (Radic et al., 1993).
Non-cholinergic Functions
The presence of AChE outside of cholinergic synapses suggests that it may be involved in functions other than the hydrolysis of ACh.
Peptidase activity has been reported to be associated with AChE from the electric eel Electrophorus electricus and foetal bovine serum (Chubb et al., 1980), and also sheep basal ganglia (Majumdar et al., 1988). Many functions have been ascribed to this activity, including the ability to cleave substance P (Chubb et al., 1980), the enkephalins (Chubb et al., 1983), and the amyloid precursor protein (Small et al., 1991). The association of AChE and peptidase activity has, however, been very controversial (Chatonnet and Masson, 1986; Checler and Vincent 1989). The trypsin-like activity of commercial E. electricus AChE has been identified as a preparative contaminant (Carroll and Emmerling, 1991),and a proteolytic activity of the cholinesterases is now largely discounted.
An HNK-1 carbohydrate epitope was discovered on AChE from electric organs of E. electricus and T. marmorata (Bon et al., 1987), whose presence had previously been used to identify cell adhesion molecules. Micro-infusion of AChE into guinea-pig substantia nigra hyperpolarises nigro- striatal dopaminergic neurones (Last and Greenfield, 1987), even after irreversible blocking with DFP or soman (Greenfield et al., 1988).
Alzheimer's Disease
A role for the cholinergic system in human memory was suggested in the early 1970s, by demonstrating that the cholinergic antagonist scopolamine (hyosine) impaired learning in man (Crow and Grove-White, 1973; Drachman and Leavitt, 1974). Whereas the AChE inhibitor physostigmine was found to increase long term memory processes (Davis et al., 1978). Around the same time it was discovered that post mortem AD brain tissue showed reduction in the cholinergic neuronal markers Choline acetyltransferase and AChE (Davies and Maloney, 1976). Loss of AChE in AD has more recently been refined to selective loss of membrane bound G4a (Schegg et al., 1992).
These findings led to the formulation of a "cholinergic hypothesis", linking abnormalities in the cholinergic system to certain functional and pathological changes in AD (Perry and Perry, 1980). Since then abnormalities have been found in other parts of the cholinergic system; ACh synthesis (Sims et al., 1980), choline uptake (Rylett et al., 1983), and in some cases, muscarinic receptors (Wood et al., 1983).
The importance of AChE in AD was emphasised by Smith and Cuello (1984), suggesting that different cell groups where lesions occur in AD share a common feature, they all contain AChE.
Cholinesterases are found in senile plaques (Friede, 1965), even at the initial stages of their formation (Morán et al., 1993), and in neurofibrillary tangles (Mesulam and Morán, 1987). Mesulam and co-workers have reported that AChE in plaques and tangles shows a lower pH optimum, a reduced sensitivity to the inhibitors BW284c51 (Geula and Mesulam, 1989), Tacrine, and physostigmine (Mesulam et al., 1987), and an increased sensitivity to indoleamine inhibitors (Wright et al., 1993). The characteristic substrate inhibition of AChE is altered in plaques and tangles, leading to the suggestion of an altered form of the enzyme (Schätz et al., 1990). NPs and NFTs present an unnatural hydrophobic environment for AChE, and it should therefore not be thought surprising that there is a change in the enzyme's kinetics. Navaratnam et al. (1991) reported an anomalous form of AChE present in the CSF in life of patients later confirmed at post mortem to have been suffering from AD. Since this anomalous form represents only a small fraction of the AChE in CSF, it has not been possible to produce any kinetic data.
Current therapeutic strategies for the treatment of AD aim mainly to alleviate cognitive deficit by activating defective cholinergic transmission. Work has mainly centred on inhibitors of AChE (Nordberg and Winblad, 1993). The only drug currently licensed in the USA for the management of AD is Tacrine, under the brand name Cognex.
To the bibliography for Cholinesterases.
*This review is essentially part of the introduction from my D.Phil. thesis, and as such I gratefully acknowledge the help of my D.Phil. supervisor Dr. J.D. Priddle for his careful proofreading and helpful suggestions.
The huge volume of work on the cholinesterases has been extensively reviewed, most notably by Silver (1974, The Biology of Cholinesterases, North Holland Publishing Co. Ltd., Amsterdam), and Massoulié et al. (1993, Progress in Neurobiology 41, 31-91), and this brief introduction in no way replaces these excellent publications.