Chung W., Boujemaa-Paterski R., Winograd-Katz S., Eibauer M., Geiger B. & Medalia O.
(2025)
BioRxiv.
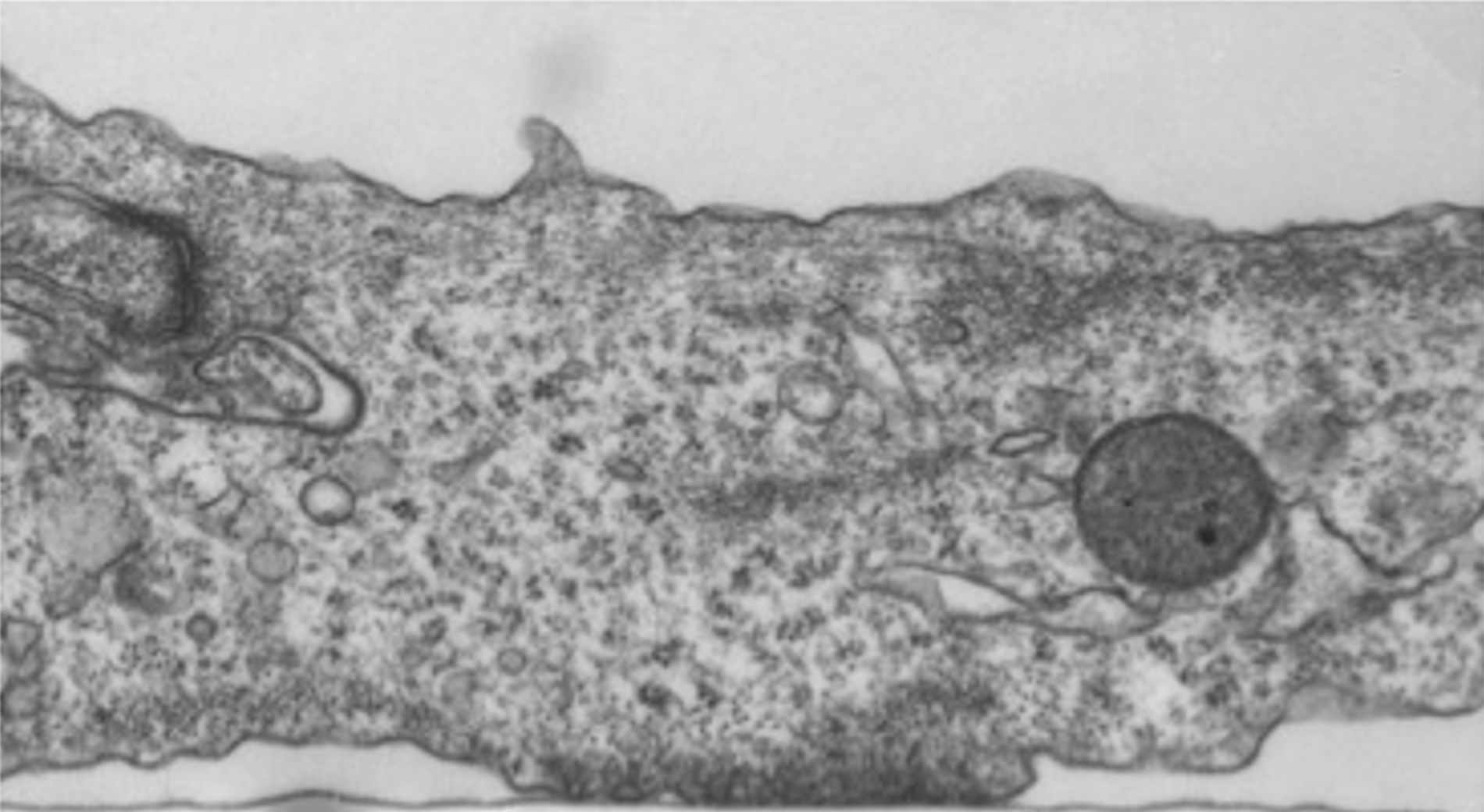
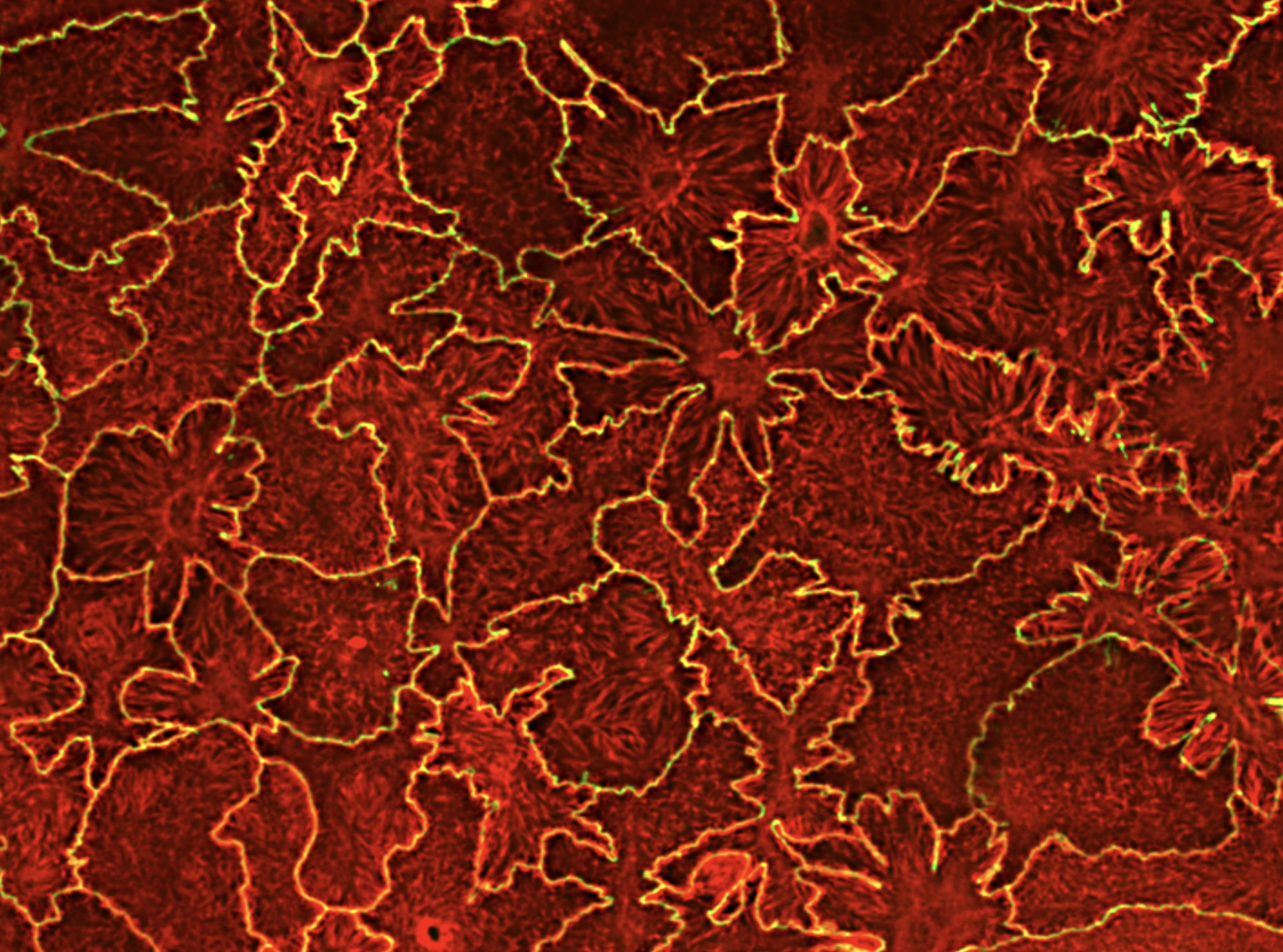
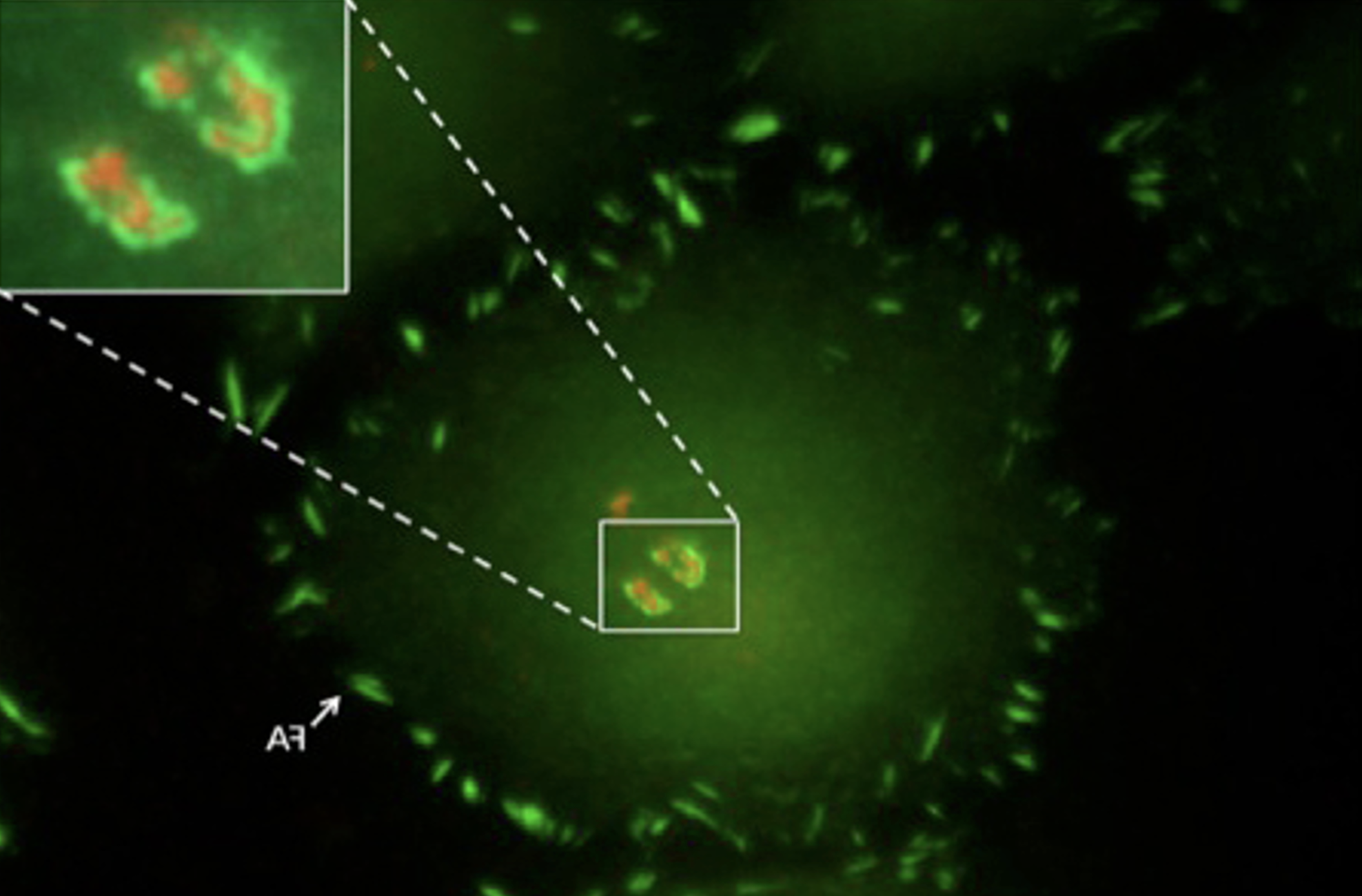
Focal adhesions (FAs) are dynamic macromolecular assemblies that anchor the actin cytoskeleton to the extracellular matrix via integrin receptors, thereby regulating cell morphology and migration. Although FA maturation and organization have been extensively studied, it remains unclear how regulatory proteins influence the 3D architecture of FAs. Here, we show that loss of the vasodilator-stimulated phosphoprotein (VASP) impairs adhesion dynamics. We employed CRISPR/Cas9-mediated knockout of VASP and/or the mechanosensitive adaptor protein zyxin to investigate their respective roles in actintextendashadhesion coupling. Loss of VASP and zyxin correlates with altered FA morphology and impaired dynamics. Using cryo-electron tomography (cryo-ET), we resolved the polarity of individual actin filaments associated with FAs and identified a contractility-related actin layer enriched with tropomyosin. VASP and zyxin are required for the assembly of dense and aligned actin bundles with uniform polarity, oriented with their barbed ends towards the cell edge. In contrast, the tropomyosin-decorated dorsal actin layer remains unaffected by these perturbations. Our findings reveal distinct, layered architectures within FAs and underscore the cooperative role of VASP and zyxin in stabilizing the organization of actin filaments at functional adhesion sites.Competing Interest StatementThe authors have declared no competing interest.
Yado S., Zoabi R., Brezinger-Dayan K., Albeck S., Unger T., Meiron M., Eisenberg G., Nahmad A. D., Gilat A. T., Besser M. J. & Geiger B.
(2025)
Frontiers in Immunology.
16,
1625118.
Adoptive T cell therapy (ACT), particularly tumor-infiltrating lymphocyte (TIL)-based therapy holds great promise for cancer treatment, yet it still faces major challenges such as patient-to-patient variability in expansion rates and cytotoxic potency. Recent studies suggest that a \u201csynthetic immune niche\u201d (SIN), composed of immobilized CCL21 and ICAM-1, can enhance both the expansion and cytotoxicity of murine and patient-derived T cells. To explore the mechanisms underlying the variability of expansion and cytotoxic potency, we conducted morphological and molecular phenotyping of TIL specimens from different donors immediately following the pre-Rapid Expansion Protocol (pre-REP) stage, enabling us to predict their expansion potential. We further developed novel SIN-based strategies that differentially reinforce the efficacy of both low- and high-expanding TILs. Our experiments revealed two distinct TIL groups with either low- or high-proliferation properties, identified across cultures derived from different patients. We further demonstrate that a 14-day REP with feeder cells and SIN facilitates the proliferation of the low-expanding cells, while the expansion of high-expanding TILs benefits from a sequential expansion protocol, consisting of 7 days with feeder cells only, followed by 7 days with SIN treatment. At the end of the REP both TIL populations display high levels of granzyme B and perforin and reduced levels of exhaustion markers. Importantly, functional cytotoxicity assays using autologous tumor targets demonstrated that SIN stimulation improved the tumor-killing capacity of low-expanding TILs, while preserving the potent cytotoxicity of the high-expanding TILs. These data indicate that the refined CCL21+ICAM1 SIN treatment improves expansion rates and activation profiles of both TIL populations, thereby enabling a powerful, personalized SIN-enhanced protocol for TIL-based immunotherapy.
Whitlock J. M., Leikina E., Wang H., Zhang W., Katz G., Reuven N., Elson A., Geiger B. & Chernomordik L. V.
(2025)
BioRxiv.
The skeleton is a living, biological tissue responding to the biomechanical demands placed upon it throughout life. The individual bones creating this physiological system are each shaped by a multinucleated cell type - the osteoclast - that sculpts each bone in collaboration with local cellular partners, which offer chemical and even tactile feedback of many sorts. Unfortunately, the perturbation of osteoclast formation and function underpins a broad range of human skeletal pathologies, including osteopetrosis - a systemic pathology characterized by impaired osteoclast resorption leading to skeletal thickening, brittle bones, frailty, and lethality. Here, we describe a molecular dysfunction observed in murine and human models of two forms of osteoclast-rich, autosomal recessive osteopetrosis, and our approach for exploiting this molecular dysfunction to correct pathologic osteoclast hyperfusion and resorptive impairment. We find that La - a manager of osteoclast fusion and subsequent resorptive activity - is greatly elevated at the surface of osteoclasts upon loss of SNX10 or OSTM1. Using inhibitory antibodies, we suppress excessive La surface function in these mutant osteoclasts, impede osteopetrotic hyperfusion and restore osteoclast resorptive function. We share these observations as proofs-of-principle that osteoclast fusion represents a viable therapeutic target for addressing osteoclast dysfunction in diseases underpinned by excessive osteoclast multinucleation and perturbed resorptive function.Competing Interest StatementThe authors have declared no competing interest.United States-Israel Binational Science Foundation, 2021168Intramural Research Program of the National Institutes of HealthEunice Kennedy Shriver National Institute of Child Health and Human Development, R00-HD110609