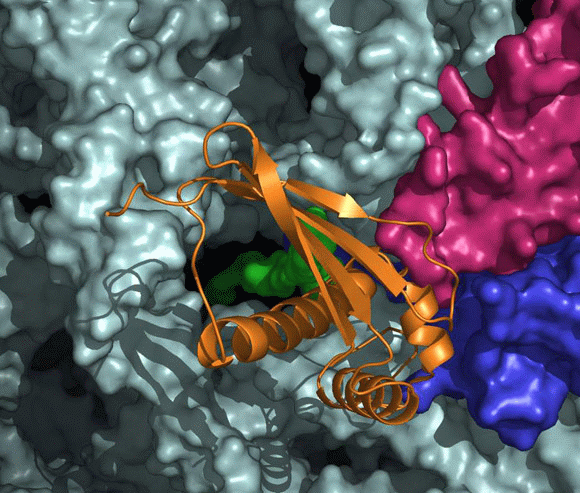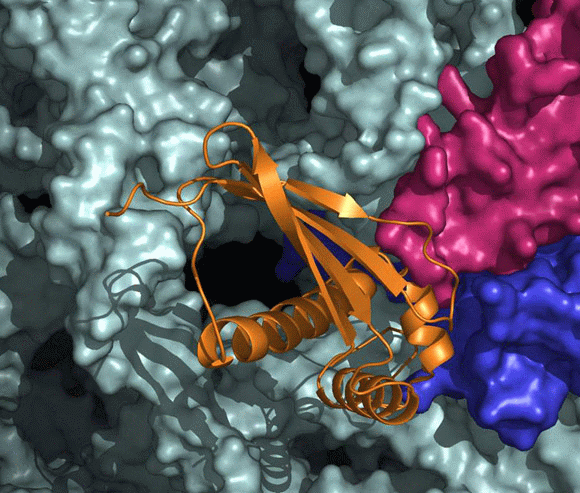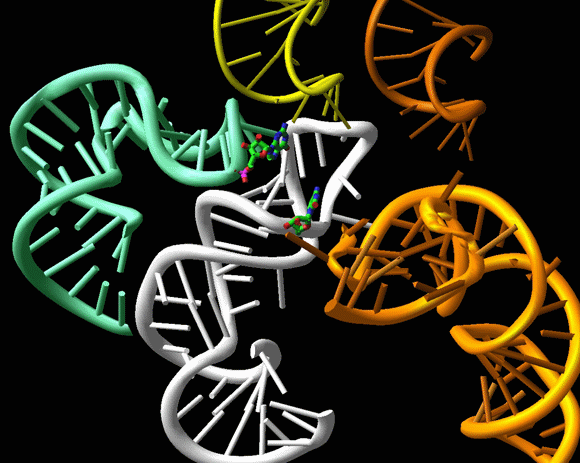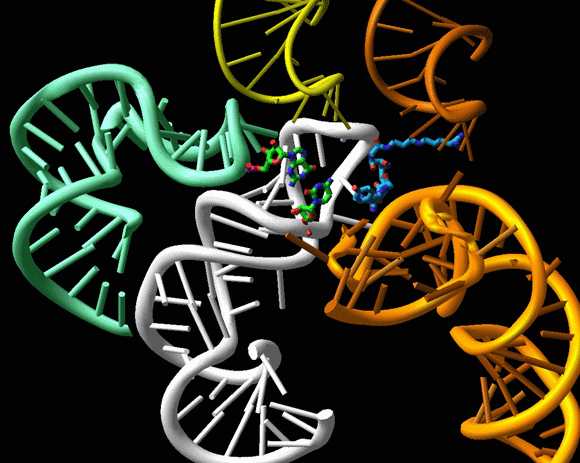
| |
Prof. Ada E. Yonath
|
Ribosome: Molecular, Structural, Functional, Evolution, Medical and Genetic aspects
We focus on the molecular and structural bases of the main aspects of ribosomal function, their inhibition (mainly be antibiotics), their origin (which provides the link between the RNA based world and the contemporary life). In parallel we are designing next generation, eco-friendly antibiotics, as well as a recombinant collagen-mRNA platform for controllable protein synthesis, alongside investigating ribosomopathies, namely the human diseases that are linked to mutations in ribosomal genes, mainly cancers and severe anemia.
BACKGROUND
The ribosome is the cellular organelle catalyzing the translation of genetic code into proteins. It is a protein/RNA assembly arranged in two subunits that associate for performing protein biosynthesis. The large subunit creates the peptide bonds and provides the path for emerging nascent proteins, and the small subunit plays a key roles in initiating the biosynthetic process and in controlling the fidelity of codon-anti-codon base pairing. The ribosome contains three tRNA binding sites, designated the A (aminoacyl), P (peptidyl) and E (exit), which are located on both subunits. During the elongation cycle both ribosomal subunits work together to translocate all three tRNAs molecules together with the associated mRNA chain by precisely one codon. In this motion each of the tRNA molecules passes through the three ribosomal binding sites, from A- to P-to E-site.
HISTORY
We initiated ribosomal crystallography about three decades ago, (Yonath et al. 1980) and determined the 3 A structures of functionally active conformations of the small and the large ribosomal subunits in 2000 and 2001 (FIGURES 1-2 and Schluenzen et al. 2000, Harms et al., 2001, Schluenzen et al 2004, Auerbach et al 2004, Yonath and Bashan 2004, Harms et al 2004 Yonath 2005, Pfister et al 2005, Yonath et al 2009, Bashan et al 2010, Bashan and Yonath 2011, Belousoff et al 2011, Zimmerman et al 2014, Eyal et al 2015, Matzov et al 2017b, Belousoff et al 2017, Eyal et al 2016, Krupkin et al 2016, Further analysis revealed the mechanism of peptide bond formation and illuminated the fashion of the ribosomal involvement in cellular regulation. In addition, by investigating crystallographically the modes of binding of over a dozen antibiotics, we elucidated the structural basis for their action, as well as for their selectivity, and illuminated possible pathways for acquiring resistance by pathogenic bacteria. Highlights of the results of our studies are described below.
NEXT GENERATION ENVIRONMENTAL FRIENDLY ANTIBIOTICS
Resistance to antibiotics is a severe problem in contemporary medicine. After identification of the common traits of the modes of action of the antibiotics targeting bacterial ribosomes in complex with these antibiotics illuminated common pathways of antibiotics inhibitory action, shown above (i.e. binding to the ribosomal functional sites), in order to illuminates the species-specific diversity in infectious-diseases susceptibility we determined structures of ribosome from a multi-resistant pathogenic bacteria. Careful comparisons of the structures of pathogenic and non-pathogenic bacteria revealed novel structural motifs, essential for protein biosynthesis but are not located in the primary ribosomal active sites, hence no mechanism for their modification that could lead to resistance are currently known. Consequently, resistance is expected to appear slowly and less efficiently. These findings prompted the design of antibiotics with desired compositions that can be optimized in terms of their chemical properties, toxicity, cellular penetration, and species-specificity, thus preserving the microbiome (that is un-intentionally damaged by the current antibiotics), as well as increasing their bio degradability, thus reducing the ecological hazards caused by the non-digestible components of the current antibiotics’ metabolites. Matzov et al 2017a, Auerbach-Nevo et al 2016
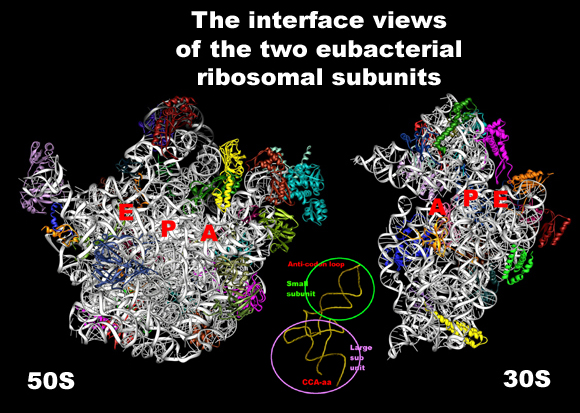
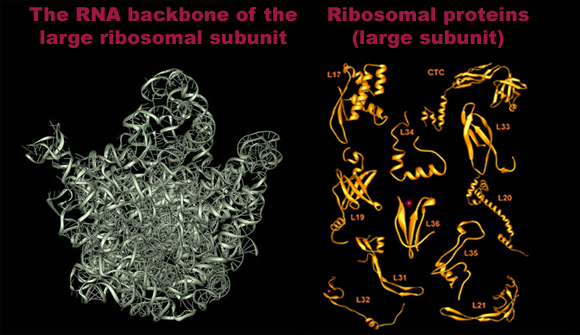
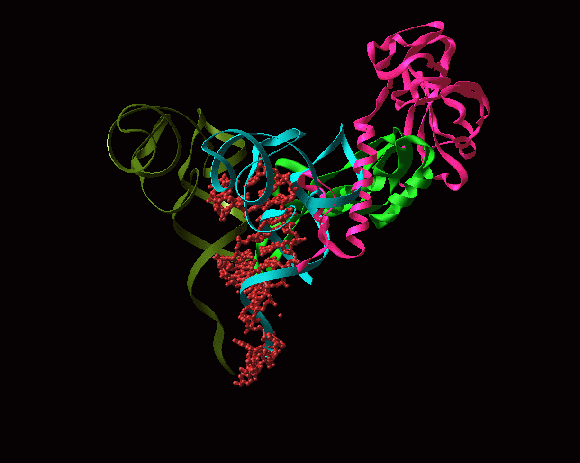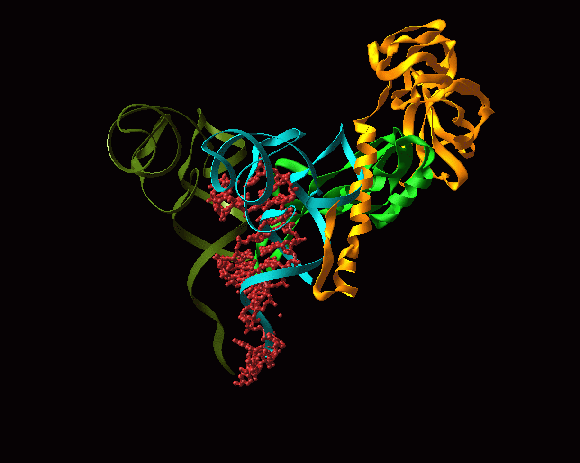
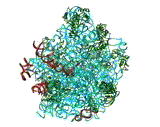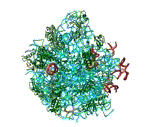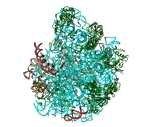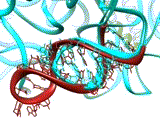
FIGURE 1: The 3 A structures of the two ribosomal subunits from eubacteria, viewed from their "front sides", namely the surfaces that create the intersubunit interfaces within the assembled 70S ribosome. Silver ribbons represent the ribosomal RNA whereas the main chains of ribosomal proteins are painted in different colors. Right: The small ribosomal subunit from Thermus thermophilus (T30S). For orientation: the "nose" and the "shoulder" are on the left side and the "platform" on the right. A, P and E designate the anticodon loops of the three tRNA molecules. Left: The large ribosomal subunit from Deinococcus radiodurans (D50S). For orientation: the L7/L12 stalk is on the right and the L1 stalk on the left. A, P and E (aminoacyl, peptidyl and exit, respectively) designate the sites of the interactions of the elbows of the three tRNA molecules with the large subunits. The Insert: A tRNA molecule (in gold) with its approximate positioning on the ribosome. The two functionally relevant feature, namely the anticodon loop and the universal aminoacylated CCA end, are marked.
FIGURE 2:
Backbone representations of the RNA (Left) and of selected proteins (Right) of the large ribosomal subunit from Dienococcus radiodurans.
A. PEPTIDE BOND FORMATION
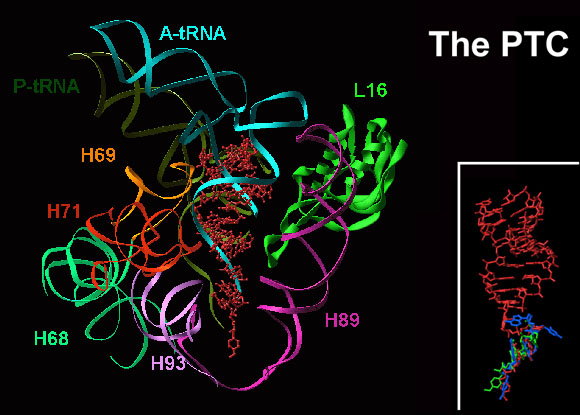
Crystallographic studied of complexes of the large ribosomal subunit with various substrate analogs, antibiotics and inhibitors confirmed that the main catalytic task of the ribosome is to provide a template for the precise positioning of tRNA molecules, rather than to participate in the actual chemical reaction. The identification of a two-fold rotation axis in the peptidyl transferase center of all known ribosomal structures led to a proposal of a unified machinery for peptide bond formation, translocation and nascent protein progression. This machinery implies that the ribosome not only positions the substrates in favorable orientation for peptide bond formation, it also guides the translocation and guaranties its accuracy.High-resolution crystal structures of large ribosomal subunits from Deinococcus radiodurans complexed with tRNA-mimics (Harms et al., 2001; Bashan et al., 2003; Agmon et al., 2003; Agmon et al., 2005 ) indicated that precise substrate positioning, mandatory for efficient protein biosynthesis is governed by remote interactions of the tRNA helical features. Indeed, superposition of precisely and not well-positioned mimics indicates marked differences in their binding modes (FIGURE 3). Thus, mimics that are not engaged in remote interactions or are somewhat misplaced mimics due to disorder of the features involved in precise placement, are positioned in the peptidyl transferase center (PTC) in conformations requiring rearrangements (Hansen et al., Mol Cell, 10, 117-128, 2002). Further analysis revealed two molecular switches: helix H69 that seems to act as a “crane” (FIGURES 4A and B) in A- to P- passage of tRNA acceptor stem, in concert with base A2602 that seems to act as a propeller (FIGURES 4C and D).We proposed a unified machinery integrating peptide-bond formation, A-to-P-site translocation, and the entrance of the nascent protein into its exit tunnel. This proposal is based on the architecture of the PTC, on the placement of tRNA mimics, and on the identification of a two-fold related region consisting of almost 200 nucleotide (over 90 nucleotide-couples) of the ribosomal RNA (FIGURE 5A), as well as on the relative orientations of the A- and P-site mimics in large ribosomal subunit from Haloarcula marismortui, H50S (Nissen et al., Science 289, 905-920, 2000; Schmeing et al., NSB 9, 225-30, 2002; Hansen et al., Mol Cell, 10, 117-128, 2002).This machinery implies peptide-bond formation in conjunction with a sovereign, albeit correlated, motions of tRNA 3' termini and involves a spiral rotation of A-site-tRNA 3'end around an approximate two-fold rotation axis that overlaps with the bond between the single stranded 3'end and the helical acceptor stem of the A-site tRNA. The sole requirement for this machinery is that the initial P-site tRNA possesses the rotated conformation.PTC features that ensure the precise orientation required for A-site nucleophilic attack on the P-site carbonyl-carbon, guide the rotatory motion (FIGURE 5A-E). The detection of similar two-fold symmetry related regions in all known structures of large ribosomal subunit indicate the universality of our proposed mechanism.
The geometrical requirement for the processivity of protein elongation is that that all A-site tRNAs are positioned accurately, so that they can undergo the rotatory motion, and that the first P-site tRNA is positioned in the so-called “flipped” configuration. The directionality of the latter is obtained since the PTC provides 2 potential base-pairs at the P-site (FIGURE 5F) instead of the single basepair at the A-site. Importantly, the two P-site base pairs allow A76 of the P-site tRNA to provide chemical catalysis to peptide bond formation (Weinger et al., Nat Struct Mol Biol 11, 1101-6, 2004).
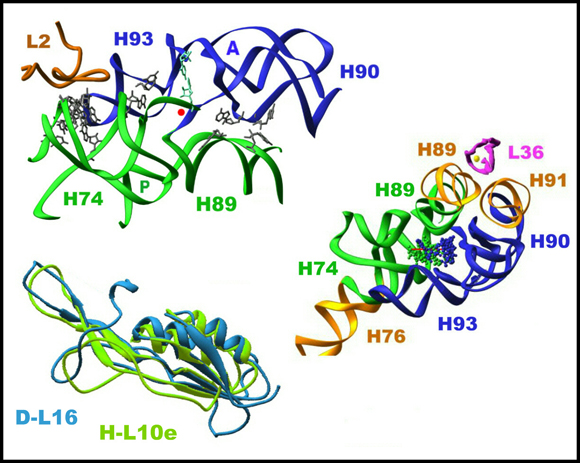
B. ON RIBOSOME EVOLUTIONThe entire symmetrical region is highly conserved, consistent with its vital function. The universality of the symmetrical region hints that the ribosomal active site evolved by gene fusion of two separate domains of similar structures, each hosting half of the catalytic activity. Importantly, whereas the ribosomal internal symmetry relates nucleotide orientations and RNA backbone fold, there is no sequence identity between the related nucleotides. The preservation of the three-dimensional structure of the two halves of the ribosomal frame regardless of the sequence demonstrates the rigorous requirements of accurate substrate positioning in stereochemistry supporting peptide bond formation. Similarly, protein L16, the only ribosomal protein contributing to tRNA positioning (Agmon et al., 2003; Bashan et al., 2003), displays conserved tertiary structure alongside diverged primary sequence (FIGURE 5A).
Although it seems that the ancient ribosome was built solely of RNA, and / or proteins were added later in order to maintain the RNA intact structure, some of those proteins contribute directly to substrate positioning and reaction processivity.
The contribution of protein L2 to the ribosomal polymerase activity may also shed some light on ribosome evolution. Protein L2 is the only protein interacting with both the A- and the P-regions (FIGURE 5G and in Agmon et al., 2005), and among its two residues involved in these interactions, one of them (229) was shown to be essential for the elongation of the nascent chain (Cooperman et al., Biochem Cell Biol 73, 1087-94, 1995). It appears, therefore, that the main function of L2 is to provide stabilization to the PTC while elongation takes place. Stabilization of the ribosomal frame is mandatory for maintaining accurate substrate positioning, required for enabling the rotatory motion, but is irrelevant to single peptide bond formation. This finding is consistent with the assumption that the ancient ribosome was made only form RNA and that the proteins were added later, in order to increase its fidelity and efficiency.
Involvement in maintaining the symmetry region architecture, and consequently in peptidyl transferase activity can also be attributed to protein L36. This small Zn containing protein (FIGURE 5G) is situated in the middle of four parallel helices and seems to stabilize their overall conformation. Two of these helices are part of the symmetry related region and two are the non-symmetrical extensions of the PTC main components. Furthermore, at its location, L36 interactions can also connect these helices with the elongation factors binding sites. Hence, in addition to stabilizing the conformation of the symmetry related region, it may also be involved in transmitting information about factor binding. The possible availability of alternative route for signaling and/or alternative means for conformation preservation, may account for the absence of L36 in some species, such as H. marismortui.
The symmetry related region connecters directly or via its extension all ribosomal features involved in protein biosynthesis (FIGURE 5H). Hence can serve as a center for transitory signals between them. Krupkin et al 2014, Huang et al 2013, Fox et al 2012, Krupkin et al 2011, Belousoff et al 2011, Bashan et al 2010, Davidovich et al 2010, Belousoff et al 2010, Massa et al 2010, Davidovich et al 2009, Agmon et al 2005, Zarivach et al 2004.
FIGURE 3:
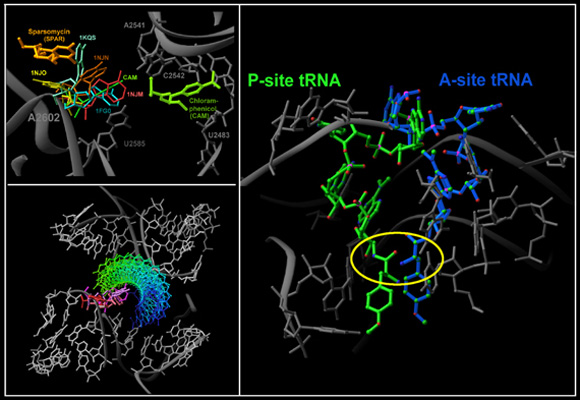
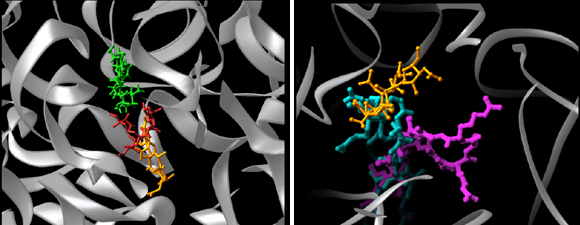
The PTC features dominating precise substrate positioning in the active site. The crystallographically determined location of a 35-mer oligonucleotide mimicking the amino acylated tRNA acceptor stem is shown in red (Bashan et al., 2003). The docked A- and P-sites tRNA (using the structure of the T70S complex, Yusupov et al., Science, 292, 883-896, 2001) are shown as ribbons, in cyan and olive-green, respectively. The insert indicates the deviations from precise positioning of the substrate when the remote contacts are not available, either because the substrate-mimic is short (blue) or because one of the main features dictating the positioning, H69, is disordered (green) (Nissen et al., Science 289, 905-920, 2000).
FIGURE 4A:A dynamic representation of the proposed role of helix H69 in translocation of A-site tRNA acceptor stem to the P-site.
Helix H69 acting as “crane” (TOP) or a “spring” (BOTTOM) for translocaing the tRNA acceptor stem from the A-(cyan) to the P-(olive green)-site. An acceptor stem mimic (Bashan et al., 2003) is shown in red. Helix H44 (light green) represents the small subunit.
FIGURE 4B:The different conformations observed for A2602 in functional complexes of the large subunit. The complexes of D50S are: ASM, an tRNA acceptor stem mimic of 35 nucleotides; ACCP: ACC bound to puromycin; SPAR: sparsomycin; CAM; chloramphenicol (Bashan et al., 2003; Schluenzen et al., 2001). The complexes of the large ribosomal subunit from Haloarcula marismortui (H50S) with short (or partially disordered) tRNA mimics are: 1FG0 (Nissen et al., Science 289, 920-30, 2000) & 1KQS (Schmeing et al., NSB 9, 225-30, 2002). Highlighted are the locations of sparsomycin and chloramphenicol. The PTC elements are shown in gray. Light: the structure of D50S in complex with chloramphenicol in dark gray, and the complex with sparsomycin in light gray.
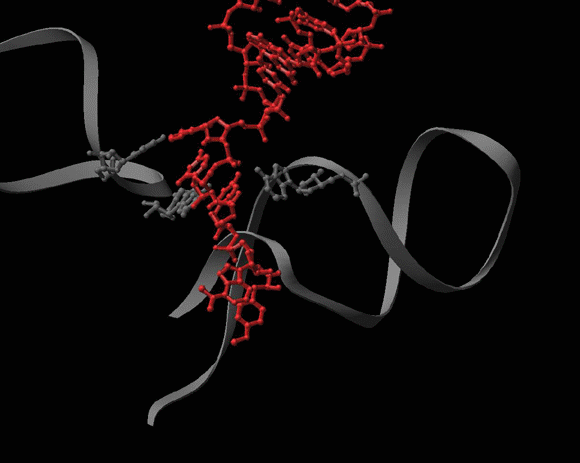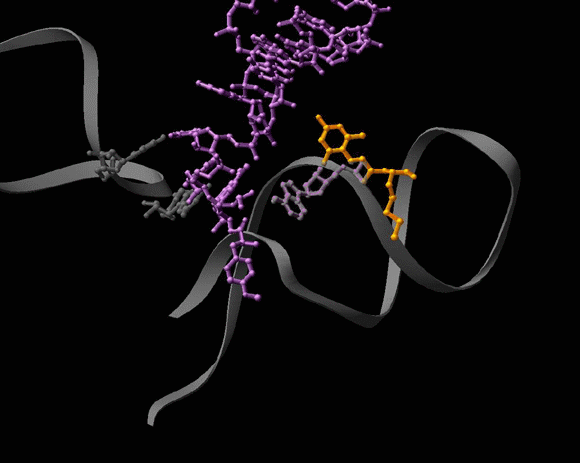
FIGURE 4C:
A dynamic representation of A2602 conformational rearrangements.
Seen is the tRNA acceptor stem mimic ASM in the absence (red) and in the presence (pink) of sparsomycin (gold). The RNA backbone in the PTC is shown is gray. A2602 is shown as a base. See also Bashan et al., 2003 and section E.
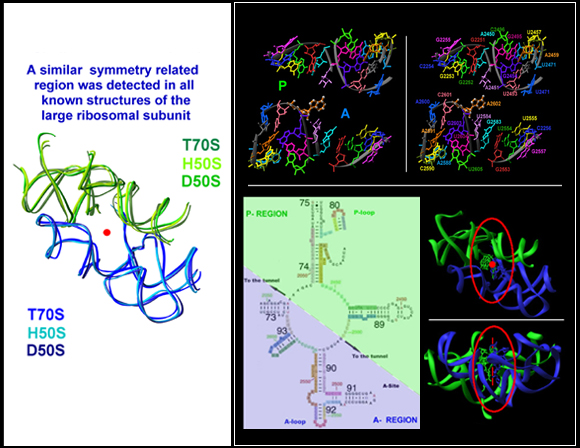
FIGURE 5A:
Left: A sizable symmetry related region was detected in all known structures of the large ribosomal subunit (the red dot represents the approximate position of two fold rotation axis).
Right: Top: A view into the active site, showing the inner shell of the symmetry related region (encircled in the right-bottom panel). The approximate locations of the A- and the P-site are marked on the left. The nucleotides are numbered on the right. Symmetry related nucleotides are colored identically. Nucleotide A2602, colored differently (in gold-silver) and breaks the symmetry, has a special role in translocation and A->P passage (see also FIGURE 4).
Bottom: Left: The two-dimensional diagram of the symmetry related region, colored in blue and green, for the A- and the P-regions, respectively. Right: The three-dimensional structure of the entire symmetry related region together with the CCA ends of the A-site (blue) and the P-site (green) 3’ ends of the tRNA molecules. The region shown on top is encircled. The red dot and rod represent the approximate position of two-fold symmetry axis, viewed from top and from the side, respectively.
& ;
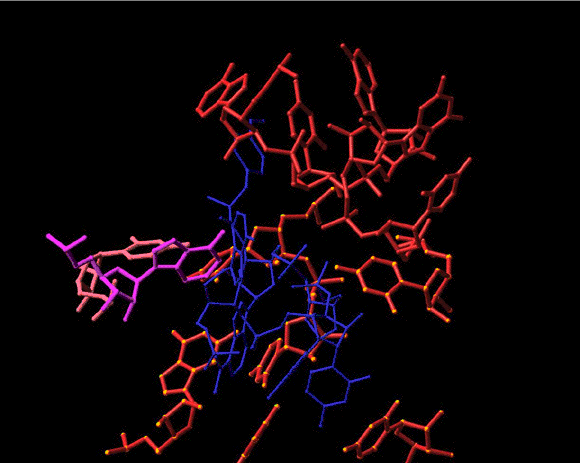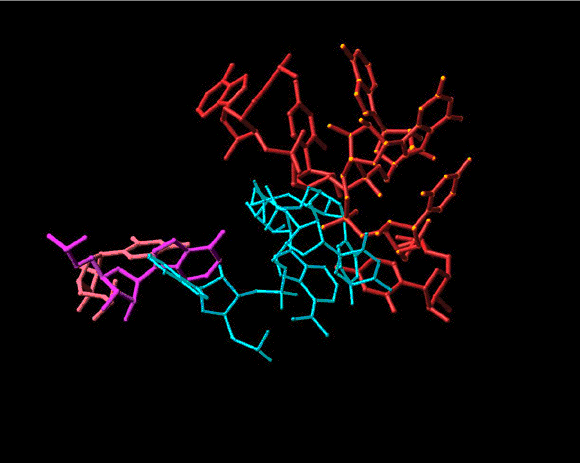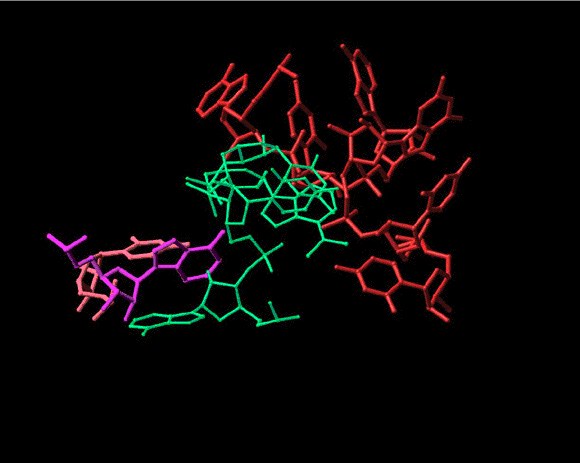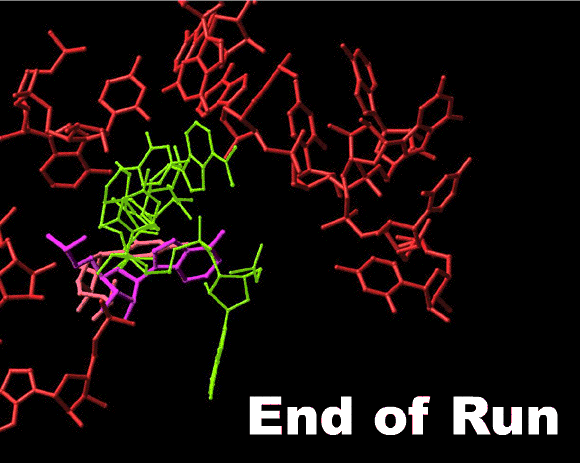
Top left: A schematic sketch portraying the basic principles of the rotatory mechanism. The tRNA 3’ ends are represented by banana shaped objects, divided by dotted lines into the four nucleotides composing them. The rear wall is drawn as ribs. The front wall anchoring nucleotides, A2602 and U2585, are not shown for clarity, but their interactions with the tRNA 3’ ends are marked by colored circles.
Top right: shows the rotating moiety (within the light-blue box), namely the aminoacylated tRNA 3’end and its connection to the tRNA acceptor stem, which translocates sidewise.
Middle and bottom: two perpendicular views of snapshots of intermediate stages in the motion of the 3’end from the A- to the P-site (gradual from blue to green). The ribosomal components are shown in gray, except for the two front-wall bulged nucleotides, which are shown in pink and magenta. The simulation of the spiral motion was performed by rotating the 3’end of the tRNA mimic (ASM, Bashan et al., 2003) in its complex with D50S by 1790 and translating it by 2Å in the direction of the tunnel.
FIGURE 5C:
Left: top: The positions of base A2602 in different 50S complexes with substrate analogs or antibiotics. Bottom: Selected A2602 positions, in relation to the rotating 3’end are shown down the two-fold axis. A- to P- passage is represented by the gradual change from blue (A) to green (P), as in FIGURE 5B.
Right: At the end of the rotation the nucleophilic amine (A-Site) and the free carbonyl (P-Site) are positioned at the stereochemistry suitable for peptidyl bond formation and for substrate – mediated chemical catalysis (Bashan et al, 2003 ; Agmon et al, 2005; Sievers et al, 2004 PNAS; Youngmanet al, 2004 Cell; Weinger et al, 2004 Nat Struct Mol Biol)
FIGURE 5D:
A dynamic representation of the rotating tRNA 3’end. In each position along the rotation, the PTC rear-wall nucleotides that guide the rotating moiety are blinking. As in FIGURE 5A, the A- to P- passage is represented by the gradual change from blue (A) to green (P).
FIGURE 5E:
The PTC features (gray) delineating the A->P site rotatory motion (blue to green)
FIGURE 5F:
The potential basepair donors (shown as atoms, within transparent bubbles) in the PTC (shown as ribbons).
FIGURE 5G:
The Bottom left: Protein L16 in eubacteria (in blue) and its equivalent in Haloarcula marismortui, called H-L10e (in light blue).
Top left: The symmetrical region (shown as ribbons), its internal contacts (shown as atoms), and protein L2,which interacts with the two sub-regions near the P-site.
Right: A different view of the symmetry region and protein L36 with the Zn atom in the middle (yellow ball).
FIGURE 5H:
The symmetry related region links the incoming and exiting tRNA molecules.
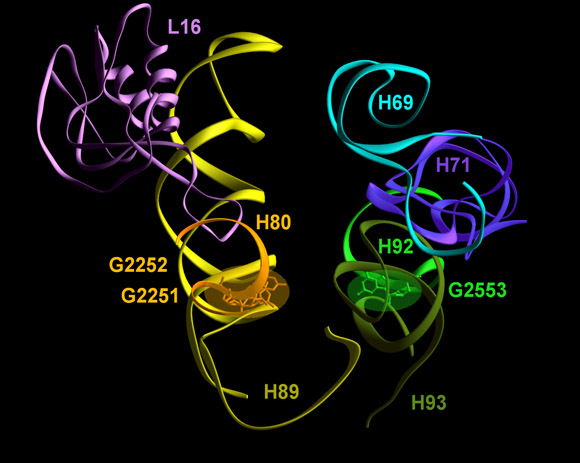
The position of the symmetry related region in the large subunit. Blue and green features represent the A- and the P-regions, respectively. The red feature, shown on top right is the intersubunit B2a bridge (H69-H71), connecting the PTC of the large subunit with the decoding site in the small subunit. The bottom left is a zoom of the view shown in top-right. The bottom right image shows the extensions of the symmetry related region (gold and pink), together with their assignments. To assist the visualization of the possible connection between the incoming and leaving tRNAs, the three docked tRNA molecules are also shown.
C. PTC MOBILITY AND ANTIBIOTICS SYNERGISM
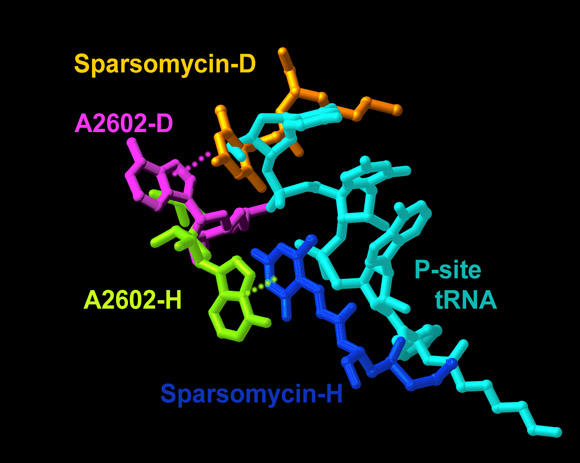
The two universally conserved nucleotides A2602 and U2585 that bulge towards the PTC center (FIGURE 5B) and do not obey the symmetry, are extremely flexible.A2602 is placed beneath A73 of A-site tRNA, within contact distance throughout the course of the rotation and exhibits a large variety of conformations (FIGURE 4A). Sparsomycin, a potent universal antibiotics agent, targets A2602 (Bashan et al., Mol Cell, 2003; Hansen et al., J Mol Biol 330, 1061-75, 2003; Porse et al., PNAS 96, 9003-8, 1999), in a fashion related to the ribosomal functional-state. By binding to non-occupied large ribosomal subunits, sparsomycin stacks to A2602 (FIGURE 6A) and causes striking conformational alterations in the PTC (FIGURE 5A), which should influence the positioning of the tRNA in the A-site (Bashan et al., Mol Cell 2003), thus explaining why sparsomycin was considered to be an A-site inhibitor, although is does not interfere with A-site substrates (Goldberg and Mitsugi, Biochem Biophys Res Commun 23, 453-9, 1966; Monro et al., Nature 222, 356-8, 1969; Porse et al., PNAS 96, 9003-8, 1999). As it faces the P-site (FIGURE 6A), it can enhance non-productive tRNA-binding (Monro et al., Nature 222, 356-8, 1969). Conversely, when sparsomycin enters the large subunit simultaneously with a P-site substrate or substrate-analog, it can cause only a modest conformation alteration of A2602, and because the P-site is occupied by the P-site substrate (FIGURE 6A), sparsomycin stacking to A2602 appears to face the A-site (Hansen et al., J Mol Biol, 330, 1061-75, 2003).
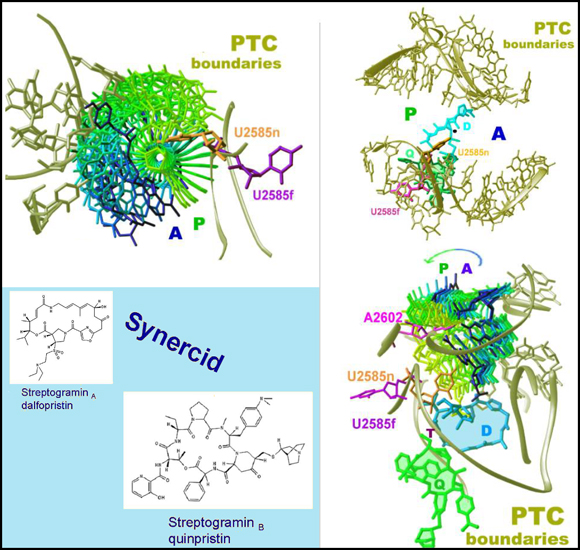
Similarly, U2585, which situated under A2602 closer to the tunnel entrance, is located within a contact distance to bound amino acid throughout the A- to P-site motion (FIGURE 6B). The base of U2585 undergoes a substantial conformational alteration (FIGURE 6B) in a complex of D50S with Synercid (FIGURE 6B), a synergetic antibiotic agent, of which one part binds to the PTC and the other blocks the protein exit tunnel (Agmon et al., 2004; Harms et al., 2004). This recently approved injectable drug with excellent synergistic activity, is a member of the streptogramins antimicrobial drug family in which each drug consists of two synergistic components (SA and SB), capable of cooperative converting weak bacteriostatic effects into lethal bactericidal activity (FIGURES 6B and C).
In D50S-Synercid complex obtained at clinically relevant concentrations, the SA component, dalfopristin, binds to the PTC and induces remarkable conformational alterations; including a flip of 180� of U2585 base hence paralyze its ability to anchor the rotatory motion and to direct the nascent protein into the exit tunnel (Agmon et al., 2004). As the motions of U2585 are of utmost importance to cell vitality, it is likely that the pressure for maintaining the processivity of protein biosynthesis will attempt recovering the correct positioning of U2585, by expelling or relocating dalfopristin, consistent with dalfopristin low antibacterial effect. The SB component of Synercid, quinupristin, is a macrolide that binds to the common macrolide binding pocket (Auerbach et al., 2004; Schluenzen et al., 2001;Yonath Bashan and 2004; Yonath 2005) Due to its bulkiness, quinupristin is slightly inclined within the tunnel, and consequently does not block it efficiently (Agmon et al., 2004; Harms et al., 2004), thus rationalizing its reduced antibacterial effects compared to erythromycin.
Since within the large ribosomal subunit both Synercid components interact with each other, the non-productive flipped positioning of U2585 is stabilized, and the way out of dalfopristin is blocked. Hence, the antimicrobial activity of Synercid is greatly enhanced. Thus, the two components of this synergetic drug act in two radically different fashions. Quinupristin, the SB component, takes a passive role in blocking the tunnel, whereas dalfopristin, the SA component, plays a more dynamic role by hindering the motion of a vital nucleotide at the active site, U2585. It is conceivable that such mode of action consumes higher amounts of material, compared to the static tunnel blockage, explaining the peculiar composition of 7:3 dalfopristin/quinupristin in the optimized commercial Synercid, although the crystal structure of the complex D50S-Synercid indicates binding of stoichiometric amounts of both components.
FIGURE 6A:
The difference between sparsomycin binding to empty D50S and to H50S bound to P-site tRNA mimic (cyan). The positions of sparsomycin in D50S and H50S are shown in gold and blue, respectively, and the corresponding conformations of A2602 are shown in magenta and yellow-green.
FIGURE 6B:
Synergism of streptogramins:
Synercid (bottom left) binding to D50S.
Top left: the conformation alteration of U2585 (1800 flip) induced by dalfopristin (SA) binding.
Right: Two orthogonal views of the positions of both dalfopristin (SA) and quinupristin (SB): down the PTC and along it, including the tunnel.
The rotatory motion is shown in top left and bottom right (Harms et al,. 2004)
FIGURE 6C:
Dalfopristin and quinupristin within the tunnel walls . All objects are shown as surfaces.
D. MORE EXAMPLES OF DYNAMIC ELEMNTS OF THE RIBOSOME
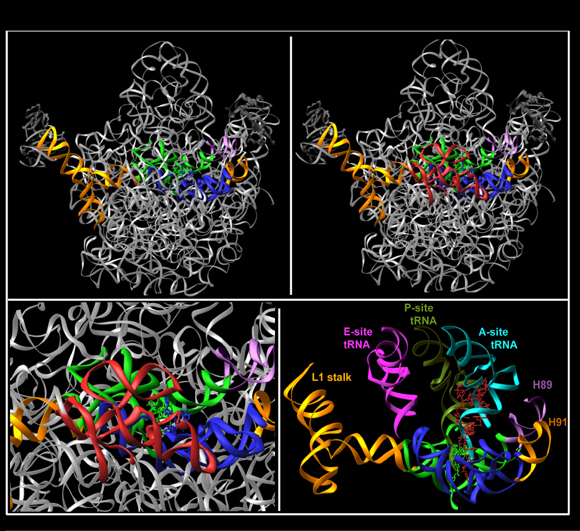
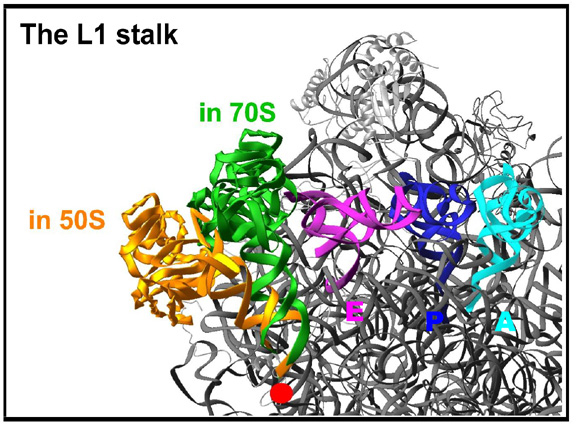
Comparative studies between unbound D50S subunit and the assembled ribosome highlighted regions with functional conformational mobility. The features are so flexible, that under specific conditions they may become disordered, as observed in the structure of the large ribosomal subunit from Haloarcula marismortui (Ban et al., Science, 289, 905-920, 2000).Among the extremely dynamic features is the L1 stalk that controls the release of the E-site tRNA (FIGURE 7A). This feature is built of helices H76-H78 (over 100 nucleotides) and protein L1.
The intersubunit bridge connecting the two ribosomal active sites: the PTC in the large subunit with the decoding region in the small one, called B2a, is built of helix H69 of the large subunit and its extension helix H70. This bridge was found to be a multi-task flexible feature:
- It connects the two ribosomal active sites, hence may transmit signals between them (FIGURES 7B and C);
- It provides many of the remote interactions required for the correct positioning of the tRNA molecule (FIGURE 3) (Bashan et al., 2003; Yonath 2003a and 2003b)
- It seems to act as a "crane" assisting the translocation of the helical stem of the A-site tRNA (FIGURE 4A) in concert with base A2602, the "propeller" of the PTC (FIGURE 4B), which participates actively in the translocation of the 3'end of A-site tRNA (FIGURES 5A-D).
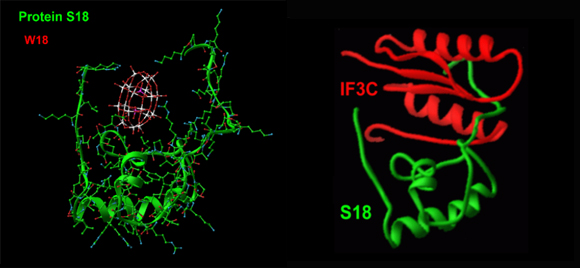
Dynamics was observed also in ribosomal proteins. Some of them undergo striking conformational changes that could be correlated to function. Proteins CTC and L22 are described below (in Sections C and D, respectively). Here we focus on protein S18. We found that the conformation of the N-terminal end of S18, S2 and the C-terminal ends of S7 and S11, all pointing into the solution, show different conformations in the two independently determined 30S structures (Schluenzen et al., 2000; Wimberly et al., Nature, 407, 327-339), indicating that these protein extensions are rather flexible. In particular, 15 residues of the N-terminal extension become ordered only upon binding of the C-terminal domain of Initiation Factor 3 (IF3) or its competitor (Pioletti et al., 2001), a cluster of 18 tungsten atoms (Schluenzen et al., 2000), called here W18 (FIGURES 7D and E). Thus, these protein extensions appear to act as tentacles, which enhance the binding and the placement of ribosomal factors like IF3.
FIGURE 7A:
The L1 stalk seems to be the feature responsible to act as the “gate" for the releasing the E-site tRNA molecules. The figure shows the left side of the upper part of D50S, as seen in FIGURE 1. The conformations of the L1 stalk in the complex of T70S with three tRNA molecules (green) and in the unbound D50S (gold) are shown. The red dot represents a pivot between the proposed “open” (gold) and “closed” (green) door conformations. The three docked tRNA molecules are also shown.
FIGURE 7B:
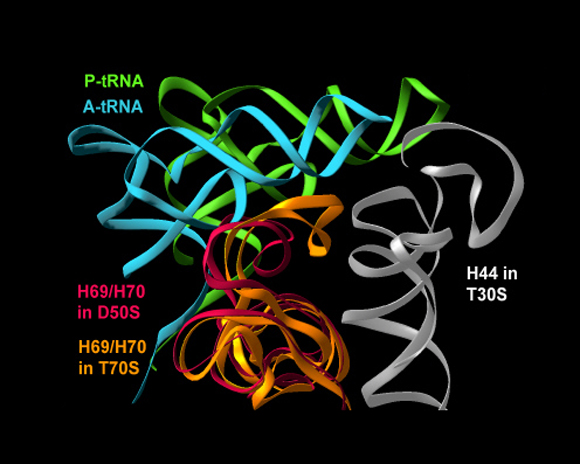
Superposition of the conformations of the B2a intersubunit bridge (H69 of the 23S RNA of the large subunit) in the unbound large subunit (red) and in the assembled ribosomes (gold). The small subunit is represented by Helix H44, which provides the decoding site and is involved in many intersubunit interactions. P- and A- site tRNAs are also shown. As seen, in the assembled ribosome P-site tRNA occupies part of the space that is used by H69 in the unbound large subunit.

FIGURE 7C:
A dynamic representation of the proposed creation of bridge B2a (H69).
When the initiation complex, reaches the large subunit to form an assembled ribosome, the P-site RNA occupies the location used by H69 in the unbound large subunit. Hence it pushes it towards the small subunit, creating the bridge connecting the decoding site with the peptidyl transferase center. The initiation complex is represented by the small helix H44 (white) together with the P-site tRNA (magenta), bound to it in the decoding center. The large subunit is represented by the B2a bridge (cyan)FIGURE 7D:
Binding of IF3(C-terminal domain) to protein S18
The conformation of protein S18 in the complexes of T30S with W18 (its competitor) and the C-terminal domain of IF3, IF3C (Pioletti et al., 2001). Note the similarity of the conformation of the N-terminal domain.FIGURE 7E:
A dynamic representation of order/disorder in S18 N-terminal extension.
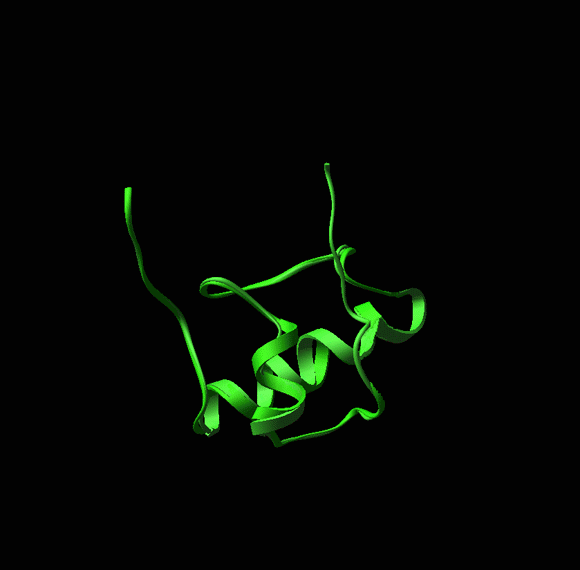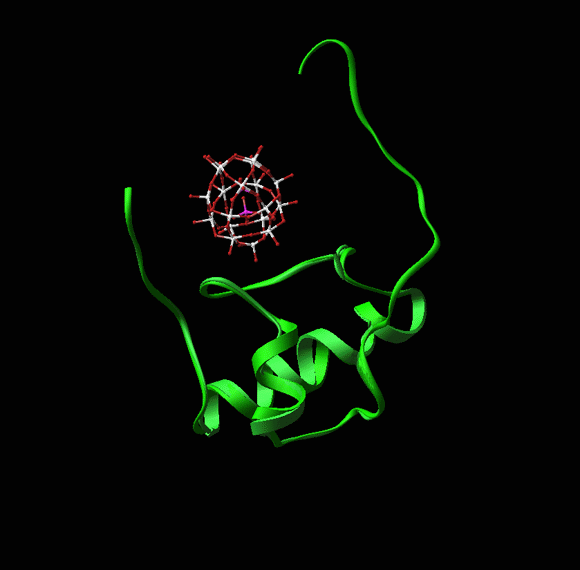
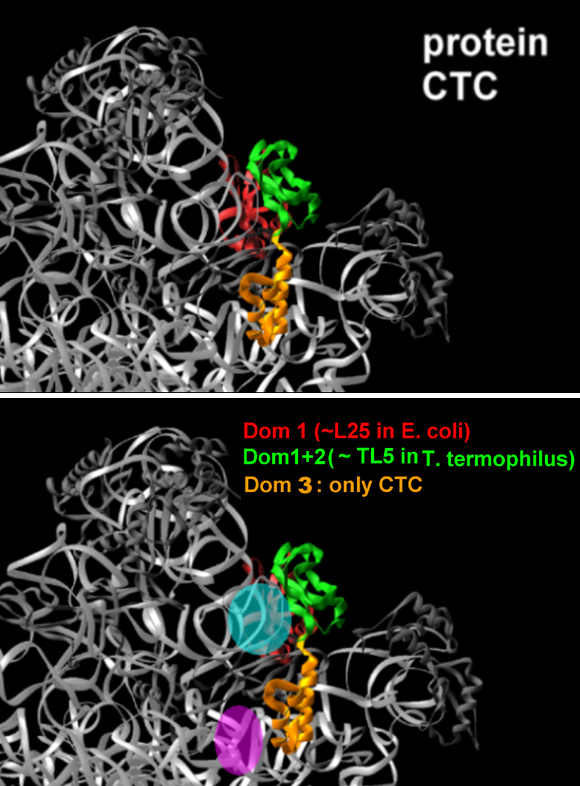
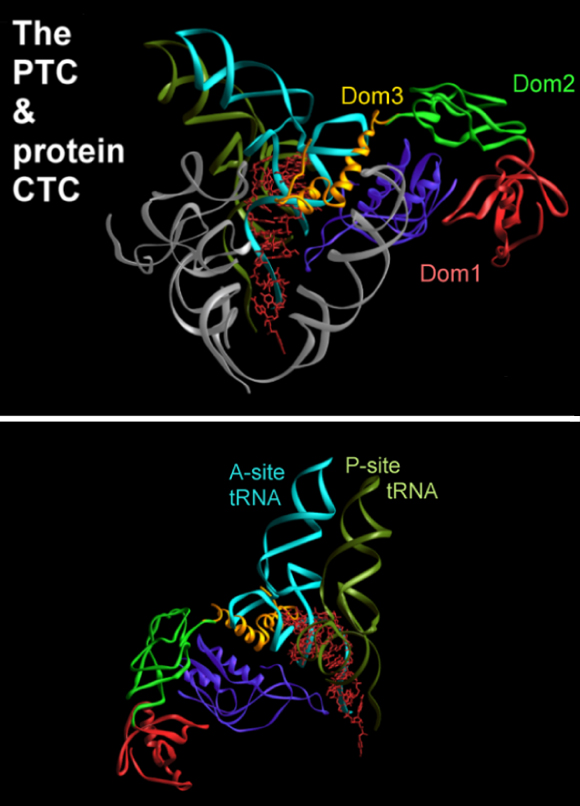
Note how W18 (the red-white-pink ball) introduces a order into the disordered N-terminal extension observed in the un-liganded structure (as in Wimberly et al., Nature, 407, 327-339).a. CTC: A RIBOSOMAL PROTEIN THAT MAY REGULATE tRNA BINDING
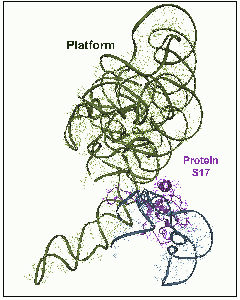
CTC, a novel D50S protein, undergoes conformational changes upon tRNA binding to D50S, although it does not interact directly with it. Protein CTC replaces the E. coli protein L25 and its T. thermophilus homologue, TL5. It contains three domains (FIGURE 8), among which the N-terminal (N-CTC) and middle domains (M-CTC) fold with a topology close to that of TL5, and N-CTC is similar to that of E. coli protein L25.In D50S N-CTC is located on the solvent side of the central protuberance, fills the space between the 5S and the L7/l12 arm, and wraps a large part of helix H38, the main constituent of the B1a intersubunit bridge, called also the A-site finger. This bridge is highly flexible, thus can readily become disordered, as observed in the structure of H50S (Ban et al., Science, 289, 905-920, 2000). N-CTC seems to protect this bridge from uncontrolled movements or interactions under mild physiological conditions. Further protection appears to be provided by M-CTC that wraps this region, probably for preventing sliding or similar motions, likely to occur at the elevated temperatures required for the growth of T. thermophilus.A similar temperature protection mechanism was detected in T. thermophilus small ribosomal subunit. Protein S17 was associated with thermal stability by genetic studies. Only in thermophilic bacteria S17 possesses a long C-terminal tail. This tail curls within a narrow groove (FIGURE 9) that separates the body from the platform, known to participate in the motions that assist translocation. In this position S17 can act as a physical stopper that blocks uncontrolled sliding of the platform that may occur at elevated temperatureC-CTC is placed at the rim of the intersubunit interface, reaching the location of the acceptor stem of A-site tRNA. Since it is connected to M-CTC by a slim linker, it may serve as an A-site binding regulator, a mechanism consistent with D. radiodurans outstanding survival. Hence, we propose a possible connection between the three domains of CTC and lives under mild, thermophilic and stressful conditions.
FIGURE 8: Protein CTC
FIGURE 8A:
A zoom into the upper part of the interface side of the large subunit, as shown in FIGUREure 1 and in the rotating image in the main home page. The backbones of the RNA and the proteins are colored gray, except for protein CTC, which is colored according to the scheme shown on the top panel. On the right side, the approximate positions of the two functional relevant features that are protected by protein CTC, are highlighted. H38, also called the B1a intersubunit bridge or the A-site finger, is shown in cyan. The position of the acceptor stem of the A-site tRNA, is shown in pink.FIGURE 8B:
Two approximately orthogonal views of the PTC and its environment in D50S, including a substrate analog (red) and the docked A- and P-site tRNAs (cyan and olive-green, respectively).
FIGURE 8C:
A dynamic representation of the conformational changes induced upon tRNA binding. The conformations of protein CTC in native D50S is shown in dark pink and in the complex with the tRNA mimic in gold.
FIGURE 9:
Protein S17 The location of protein S17 between the platform and the lower part of the small ribosomal subunit.
b. TUNNEL GATING AND CELLULAR REGULATION
Stretching from the site of peptide bond formation to the other side of the large ribosomal subunit, the exit tunnel provides the path of emerging nascent proteins. Being of utmost importance, this tunnel is targeted by many macrolide antibiotics, which bind to a specific pocket and hamper nascent protein progression.
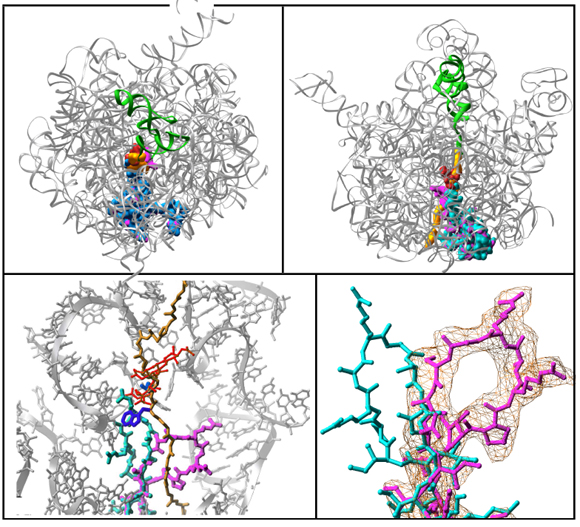
FIGURE 10A:
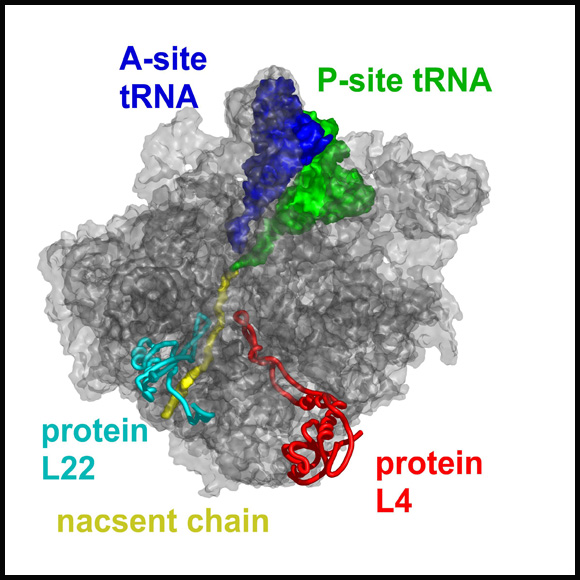
A semi transparent (gray) representation of halved large ribosomal subunit from D. radiodurans (D50S), as determined crystallographically. A modeled nascent chain (in yellow) marked the approximate position and shape of the tunnel. Proteins L4 and L22 are shown since they form the tunnel constriction.
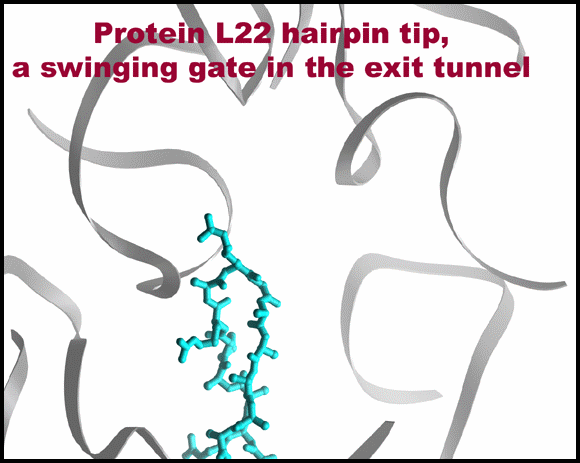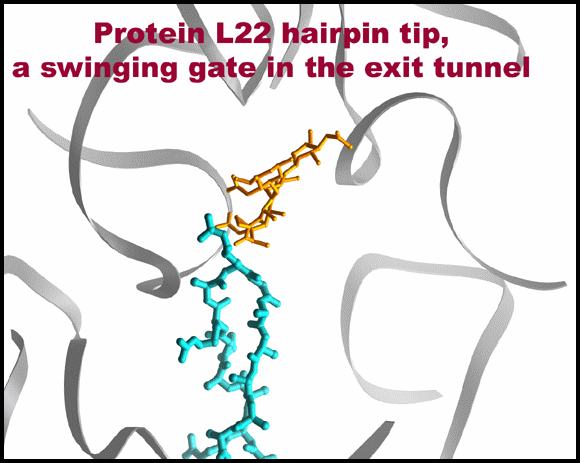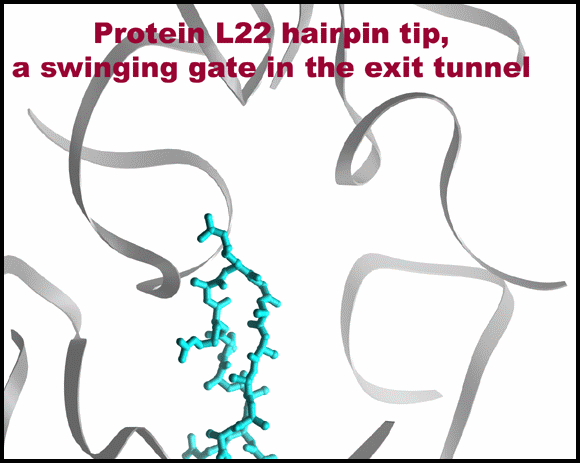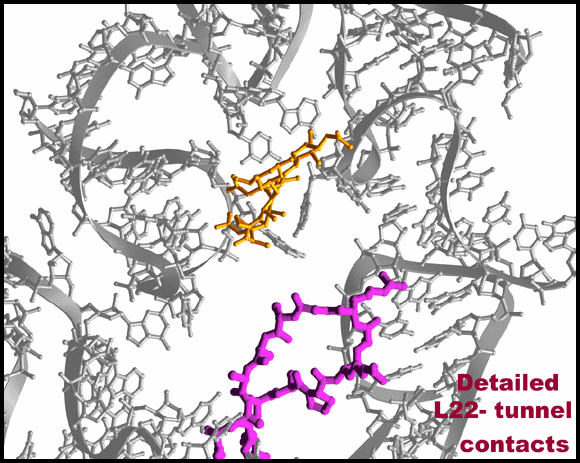
Protein L22 tip flipping across the protein exit tunnel.
The L22 hairpin tip is shown in its native (cyan) and flipped (magenta) conformations. TAO is shown in gold, except for in 10C & D where is it shown in red. Ribosomal components are in gray. A modeled poly-proline nascent chain is shown in gold. The side chain of the two residues required for SecM arrest (Nakatogawa and Ito, 2002 Cell, 108, 629-636) are highlighted in blue (Ile and Trp), whereas the essential Pro (side chain not shown) is located at the PTC (the top of the nascent chain).TOP:
A dynamic representation of the flipping tip, viewed perpendicular to the tunnel’s long axis.
BOTTOM:
A dynamic representation of the flipping tip, viewed at the entrance to the tunnel.
FIGURE 10C:
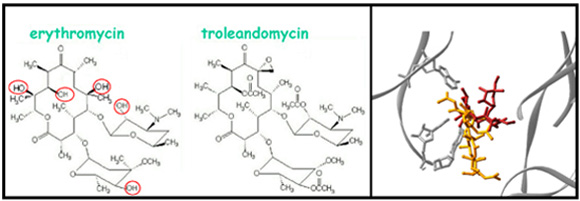
TAO’s chemistry. Left: The chemical compositions of erythromycin (see section E and FIGURE 11) and TAO. Hydroxyls existing in erythromycin, but not in TAO are circled. Right: The positions of erythromycin (red) and TAO, viewed perpendicular to the tunnel’s long axis.
FIGURE 10D:
The correlation between TAO binding and elongation arrest
Top: The entire rRNA of the 50S subunit, viewed from the entrance to the tunnel (Left), and from the interface side (Right). The entire protein L22 is shown, with its native structure in cyan, and the flipped conformation in magenta. The modeled SecM arrest sequence is shown in gold, TAO in red and P-site tRNA in green. The Bottom symbolizes our proposed mechanism for tunnel arrest. It shows a superposition of TAO (yellow) and the modeled SecM arrest sequence (blue) in the tunnel (perpendicular view), highlighting (in Red) the positions of the crucial residues for ribosomes arrest (Trp and Ile in red). The Left panel shows the electron density difference map, of the swung conformation in D50S/TAO.
FIGURE 10E:
TAO in the tunnel
Left: The position of TAO (gold) in the tunnel, compared to erythromycin (red) and a modeled 5-residues peptide (green). Right: A diagonal view into the tunnel, showing TAO (gold) and the flipped L22 hairpin tip in the native (cyan) and flipped (magenta) conformations.
E. THE ENCOUNTER OF THE NASCENT CHAIN WITH THE FIRST CHAPERONE: THE TRIGGER FACTOR
Although, in principle, protein can fold with no assistance of additional factors, since their sequences entail their unique folds, under cellular conditions nascent polypeptides emerging out of the ribosomal tunnel are prone to aggregation and degradation, and thus require assistance. The cellular strategy to promote correct folding and prevent misfolding involves a large arsenal of molecular chaperones.
Trigger factor (TF), a unique feature of eubacteria, is the first chaperone encountering the emerging nascent chain. This 48 kDa modular protein is composed of three domains, among which the TF N-terminal domain (TFa) that contains a conserved "signature motif", mediates the association with the ribosome. It cooperates with the DnaK system, and their combined depletion causes a massive aggregation of newly synthesized polypeptides as well as cell death above 30�C (Deuerling et al., Nature 400, 693-6, 1999).
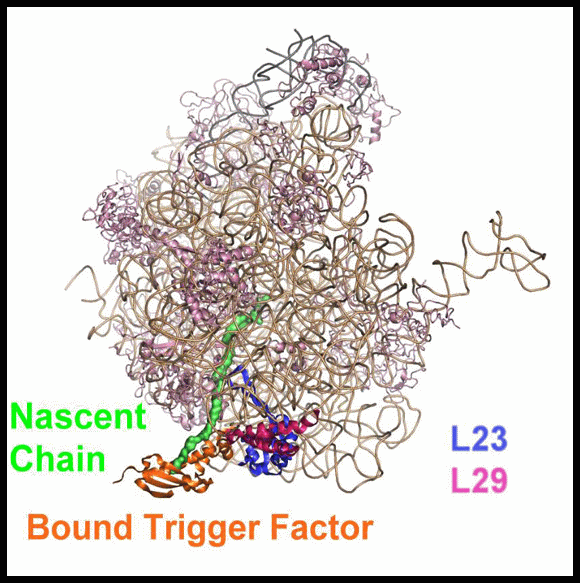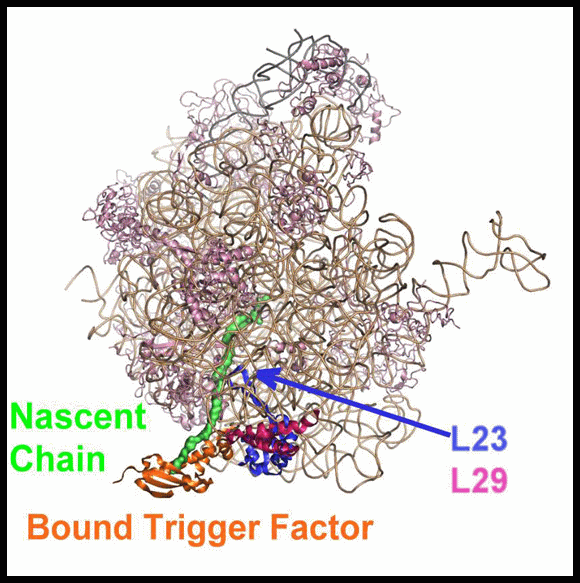
The high resolution crystal structure of D50S in complex with TFa from the same source shows that TFa binds (FIGURES 11A and B) at a triple junction between ribosomal proteins L23 and L29 and the 23S rRNA (Baram et al., 2005), and indicates that upon binding the TFa undergoes significant conformational rearrangement (FIGURE 11C). These alterations result in the exposure of a sizable hydrophobic patch facing the interior of the ribosomal exit tunnel, which should increase the tunnel's affinity for hydrophobic segments of the emerging nascent polypeptide. Thus, the trigger factor prevents aggregation of the emerging nascent chains by providing a competing hydrophobic environment (Baram et al., 2005).
In eubacteria, protein L23 posses a unique feature, an elongated hydrophobic loop, located at the wall of the ribosomal tunnel (FIGURES 11A and B). This loop may be available for interactions with the progressing nascent chain; hence it can be involved in co-translational folding. It seems, therefore, that protein L23 plays multiple roles in eubacteria. It is essential for the association of TF with the ribosome, and may control the pace of the entrance of the nascent chain into its shelter, hence serving as a channel for cellular communication with the nascent chain while progressing in the tunnel.
FIGURE 11A:
A side view of the large ribosomal subunit. The rRNA and r-proteins (except L23 and L29) are shown as ribbons in brown and dark red, respectively. The bound trigger factor binding domain (TFa) is shown in orange, ribosomal proteins L29 and L23 in magenta and blue, respectively, and the modeled polypeptide chain in green. Note the elongated loop of L23, a unique eubacterial feature, which reaches the interior of the tunnel, to a location allowing its interaction with the emerging nascent chain.
FIGURE 11B:
A “surface representation” of the tunnel opening in the large ribosomal subunit. The rRNA and r-proteins (except L23 and L29) are shown as ribbons in brown and dark red, respectively. The bound trigger factor binding domain (TFa) is shown in orange, ribosomal proteins L29 and L23 in magenta and blue, respectively, and the modeled polypeptide chain in green. Note the elongated loop of L23, a unique eubacterial feature, which reaches the interior of the tunnel, to a location allowing its interaction with the emerging nascent chain.
FIGURE 11C:
Conformational changes upon trigger factor binding to the large ribosomal subunit.
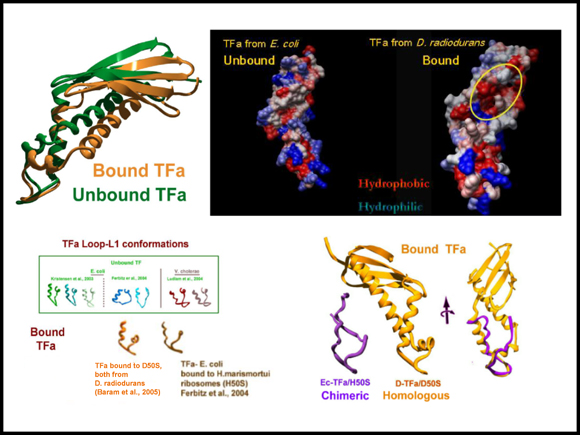
Top: the trigger factor binding domain (TFa) in the bound and unbound states. The right image shows the hydrophobic region that becomes exposed upon binding.
Bottom: the conformational variability of the TFa binding motif (called L1 loop) in unbound and bound states of the homologous (D. radiodurans) and chimeric (50S from Haloarcula marismortui with TFa from E. coli) complexes. The right side images show the entire TFa domain in the homologous complex in comparison to the region that could be resolved in the chimeric complex (Ferbitz et al., Nature 431, 590-6).
F. ANTIBIOTIC TARGETING RIBOSOMES
Resistance to antibiotics is a major problem in modern therapeutics. Antibiotics are natural or man-made compounds, designed to interfere with the metabolism or microorganisms and eliminate them by inhibiting fundamental cell processes.Ribosomes of pathogenic bacteria provide a target for many antibiotics. The two crystallizable ribosomal subunits from eubacteria, T30S and D50S, were found suitable to serve as pathogen-models (Pioletti et al., 2001; Schluenzen et al., 2001; 2003; Bashan et al., 2003; Berisio et al., 2003a; Berisio 2003b; Schluenzen et al., 2004; Harms et al., 2004; Auerbach et al., 2004; Yonath and Bashan 2004; Yonath 2005) Analysis of high resolution structures of complexes of antibiotics with these ribosomal particles shed light on antibiotic selectivity and illuminated various modes of their action, from reducing of decoding accuracy, via limiting conformational mobility, to interference with substrate binding and hindrance of the progression of growing proteins. We found that the antibiotics bind primarily to ribosomal RNA and in many cases they do not cause major conformational changes. Only a few agents trigger significant allosteric alterations, among them is sparsomycin. This universal inhibitor triggers significant conformational alterations in both the A- and the P-sites, since it forms stacking interactions with A2602, shown to be a highly mobile nucleotide (FIGURES 3, 4B and 7D)a. SMALL SUBUNIT ANTIBIOTICS
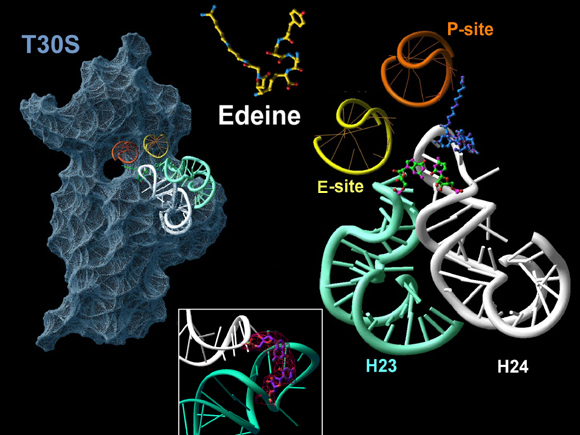
Aminoglycosides are known to render the translational apparatus highly inaccurate. They bind adjacent to the decoding site and influence its orientation. Tetracycline was found to be a multi-site antibiotic with inhibitory action that stems from its interference with A-site tRNA binding. Consistently, this binding site is also in close proximity to mutations in 16S RNA that acquire tetracycline resistance. Edeine, a universal agent, inhibits the initiation of protein synthesis by linking critical features for tRNA, IF3 and mRNA motions. It also induces an allosteric base pair between the major helices of the platform, hence limiting its conformational mobility and influencing the positioning of P-site tRNA as well as hampering the regulatory action of IF3. Thus, edeine exhibits a novel mode of action. It prevents adopting conformations required for ribosomal function by binding to them, and induces an allosteric change, the formation of a rather remote new base pair, an important principle of antibiotic action (FIGURE 12).
FIGURE 12:
Edeine, a universal small subunit antibioticsFIGURE 12A:
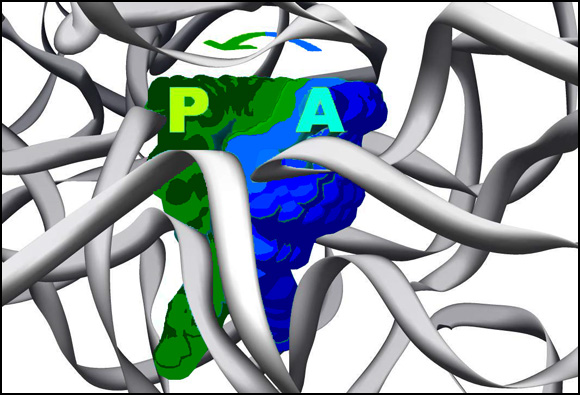
The binding site of edeine, the universal antibiotic agent, which inhibits the initiation of protein synthesis by linking together critical features of the ribosomal platform, H23 and H24 of the 16S RNA. The mobility of these components is needed for translocaing the mRNA/tRNA. The small subunit is shown as an opaque gray body with the mRNA channel in the middle of the “neck”, from a view diagonal to that of FIGUREure 1 and of the rotating image on the main home page. The chemical structure of edeine is also shown on top. The bound edeine (blue) causes an allosteric conformational change, forming a new base-pair (green) between helices H23 and H24. The electron density map of this new base pair is shown in the insert.
A dynamic representation of edeine binding and the allosteric rearrangements it creates - the new base pair.
Color code as in FIGURE 12A
b. LARGE SUBUNIT ANTIBIOTICS:
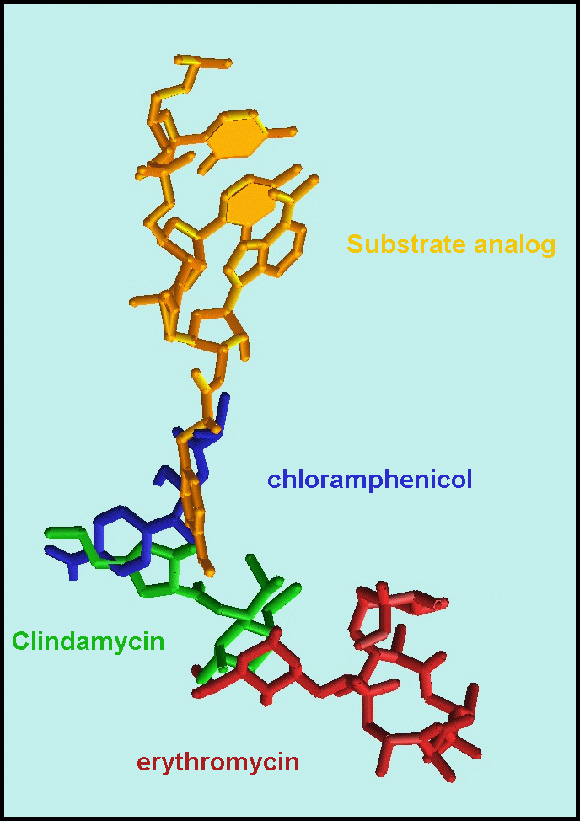
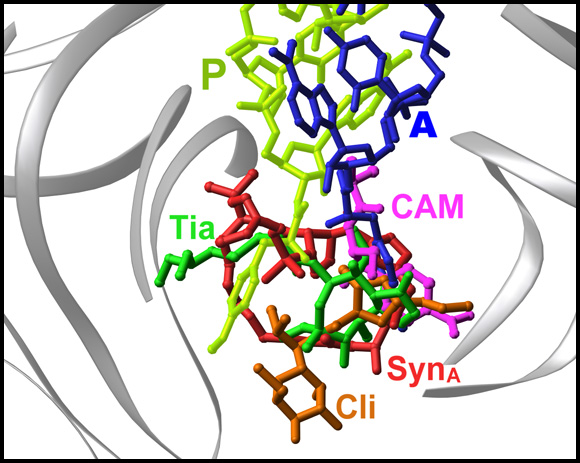
1. MACROLIDES AND KETOLIDES AND PTC ANTIBIOTICSMost of the clinically useful antibiotics target the large subunit in the PTC, its vicinity or at the entrance to the ribosome tunnel (FIGURES 13A-C). Structurally, the antibiotics that target the PTC can be divided to two groups. The members of the first group block substrate binding or interfere with the formation of the peptide bond. The members of the second group hinder either the PTC or the substrate motions. Chloramphenicol, clindamycin, tiamulin, sparsomycin, and streptograminA target the PTC (FIGURE 13B) (Bashan et al., 2003; Harms et al., 2004), and despite the limited space and the apparently similar outcome of their binding, each of these compounds acts in a different way. Chloramphenicol blocks only the A site, whereas clindamycin, tiamulin, and streptograminA bind to both the A and the P sites. Also, marked differences have been observed in the interplay between their binding and the PTC conformation. Whereas chloramphenicol and clindamycin cause hardly any conformational modifications, tiamulin induces subtle changes, and sparsomycin (Schluenzen et al., 2004) and streptograminA (Harms et al., 2004) act by triggering substantial alterations.
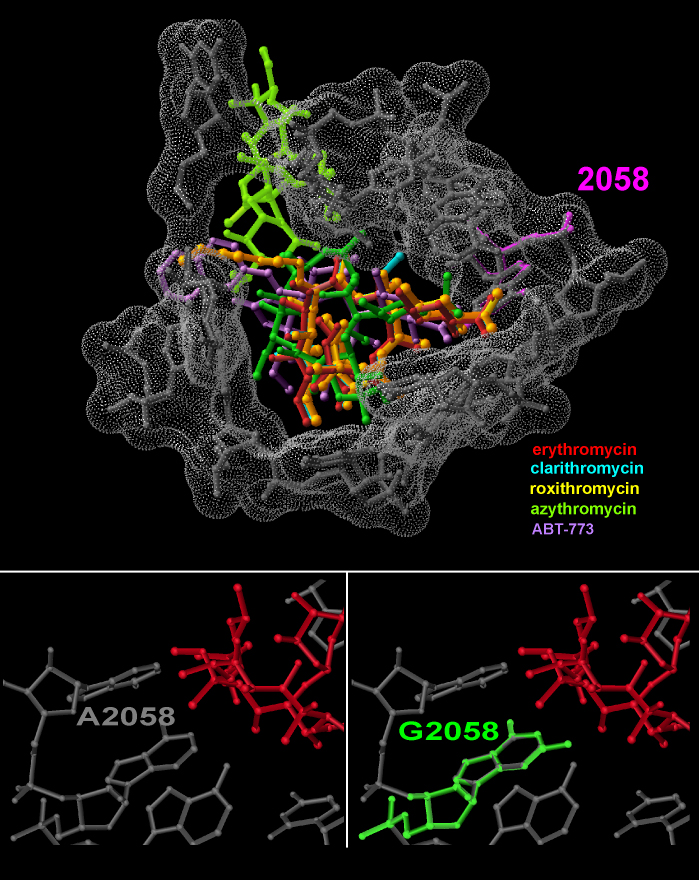
Ketolides belong to an advance class of the macrolide family, designed to overcome macrolide resistance, caused by A2058 methylation or its mutation to G . They are characterized by a keto group at position 3 of the macrolactone ring, a single aminosugar moiety and an extended hydrophobic arm. As macrolides, the ketolides bind to a specific pocket in the eubacterial tunnel, and act by producing a steric blockage of the ribosome exit tunnel and hamper the progression of nascent chains. Different from macrolides, which bind mainly to one side of the tunnel, the extended arms of the ketolides reaches the other side of the tunnel (FIGURE 13C). In this way they compensate for the loss of binding affinity to the increased bulkiness of A2058 (Schluenzen et al., 2003; Berisio et al., 2003 J Bacteriol).
The macrolides, antibiotics containing a central lactone ring of 12-16 members, block the progression of the exiting nascent chains. Nucleotide A2058 is the key determinant for binding and selectivity consistenty 14-member macrolide resistance is acquired by A->G mutation in A2508 or its methylation action. The 15- and 16-member macrolides are less selective. (FIGURE 13C). However extremely high concentrations of antibiotics are needed for their binding. Importantly, macrolides that do bind to large subunits possessing Guanine in position 2058, are oriented in conformations that are less effective for the blockage of the nascent chain progression (FIGURE 14B).
FIGURE 13A:
Antibiotics overlap of tunnel blockers, represented by erythromycin (red) with A-site PTC, represented by chloramphenicol (blue), via clindamycin (green), which binds between A-and P-sites and extends into the tunnel.FIGURE 13B:
PTC antibiotics
The binding modes of chloramphenicol (CAM), clindamycin (CLI), tiamulin (TIA) and streptograminA (SynA). The ribosomal components are shown in gray and A- and P-sites tRNAs are in blue and green, respectively).
2. MUTATION ENABLES MACROLIDES BINDING:
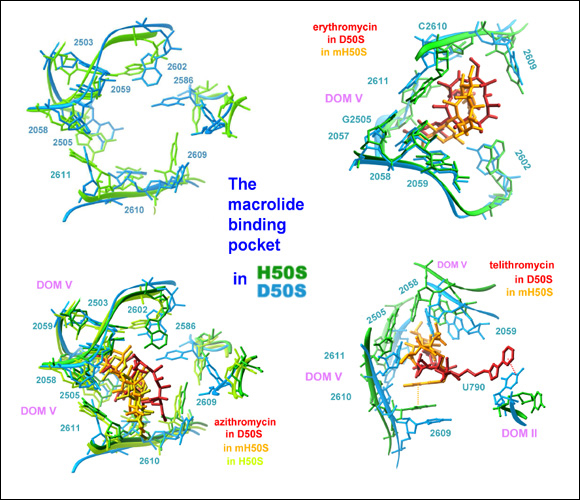
Crystal structures of the macrolides bound to the large ribosomal subunit from Deinococcus radiodurans, D50 (Schluenzen et at, 2001), a eubacterium resembling pathogens, the archaeon Haloarculla marismortui, H50S (Hansen et al, 2002 Mol Cell), which shares properties with eukaryotes, and its single base mutant G2058A (Tu et al, 2005 Cell) (called MH50S) allowing macrolide binding, indicated that despite the overall conservation of the ribosome, subtle variations dominate antibiotics selectivity, thus facilitating their therapeutical usage. It also showed the difference between binding and antibiotics effectiveness.
G2058 in H. marismortui 23S rRNA has recently been mutated to an adenosine (Tu et al., Cell 121, 257-270, 2005). This mutation (called below mH50S) increases macrolide binding affinity by 10000-fold, but did not significantly improve the effectiveness of the binding mode, as the magnitude of tunnel blockage in mH50S remained lower than that achieved by the same drug in the eubacterial D50S (FIGURE 14B). This finding indicates that although 2058 identity determines whether binding occurs, the conformations and the chemical identities of the other nucleotide in the macrolide-pocket govern the antibiotics binding-modes and, subsequently, the drug effectiveness.
Interestingly, all mH50S bound macrolides/ketolides share a similar conformation, which is almost identical to that suggested by NMR studies to be of the lowest free energy at ribosome-free environments. These ribosome-free experiments ignored the ribosome, which by providing a significant interaction network, alters radically the drug environment. Hence, the preservation of conformation of the drug in isolation is inconsistent with the high binding affinities between ribosomes and macrolides/ketolides.
The case of telithromycin-mH50S complex (FIGURE 14B) shows the separation between binding and effectiveness, since in mH50S telithromycin does not create the prominent interactions of ketolides with domains II (FIGURE 14). Likewise, the similarity between the binding modes of telithromycin and erythromycin is inconsistent with profound differences detected between the susceptibility of A2058G ribosomes to ketolides (Pfister et al., 2005).
The rational behind the strange properties of the macrolides/ketolides binding modes to mH50S may be linked to the high salinity (> 2.5 M KCl) essential for H. marismortui optimal growth and for maintaining its integrity (Shevack et al., 1985; Yonath, 2002). It is conceivable that in addition to the phylogenetic and conformational variability between archaea and eubacteria, which leads to dissimilarities between antibiotics conformations in D50S vs. H50S-mH50S (Baram and Yonath, 2005; Pfister et al., 2005), the high salinity within the H50S and mH50S masks potential ribosomal entities that could have interact with the drug.
To conclude, the G -> A Mutation of 2058 in H. marismortui ribosome was found to be most beneficial for ribosomal-antibiotics research. It confirmed that 2058 is the key player in macrolide binding; it clarified the distinction between mere binding and antibiotics inhibitory effectiveness; and it provided structural insight into the intriguing question, which is also of utmost importance for drug development: What is the relevance of "minimum free-energy conformation" determined in ribosome-free environment to antibiotics binding and their therapeutic effectiveness? In other words: is there a correlation between the "minimum free-energy conformation" of a drug, determined in ribosome-free environment and its therapeutic effectiveness?
FIGURE 14A:
The locations of several macrolides, an azalide and a ketolides within a section perpendicular to the long axis of the protein exit tunnel. The tunnel wall is shown as a semi-space-filled space, with the positions of the relevant nucleotides. A2508, a key contributor to the selectivity of and the resistance to macrolides, is highlighted in pink.
Bottom-left: Erythromycin (red) and 2058, as observed in D50S/erythromycin complex.
Bottom-right: Guanine (green) has been modeled instead of adenine 2058.
FIGURE 14B:
Macrolides selectivity and binding (H50S, mH50S and D50S as targets)
Superposition of the macrolides binding pockets in the eubacterial ribosome (50S, in blue) and that of the archaeon H50S (green). Note the phylogenetic and conformational differences, which cause less effective binding in H50S (namely a smaller portion of the tunnel is blocked in all cases).
Also note (bottom left) that there is hardly any difference in the binding mode to native H50S and to the G2058A mutant, although the binding affinity to the latter is 106 higher than to the native halophilic ribosome.
Of particular interest is telithromycin, a ketolide known to reach domain II in eubacteria (including all pathogens), which binds (in a folded state) to domain V only in the archaeon H50S.
NEXT GENERATION ENVIRONMENTAL FRIENDLY ANTIBIOTICS
Resistance to antibiotics is a severe problem in contemporary medicine. After identification of the common traits of the modes of action of the antibiotics targeting bacterial ribosomes in complex with these antibiotics illuminated common pathways of antibiotics inhibitory action, shown above (i.e. binding to the ribosomal functional sites), in order to illuminates the species-specific diversity in infectious-diseases susceptibility we determined structures of ribosome from a multi-resistant pathogenic bacteria. Careful comparisons of the structures of pathogenic and non-pathogenic bacteria revealed novel structural motifs, essential for protein biosynthesis but are not located in the primary ribosomal active sites, hence no mechanism for their modification that could lead to resistance are currently known. Consequently, resistance is expected to appear slowly and less efficiently. These findings prompted the design of antibiotics with desired compositions that can be optimized in terms of their chemical properties, toxicity, cellular penetration, and species-specificity, thus preserving the microbiome (that is un-intentionally damaged by the current antibiotics), as well as increasing their bio degradability, thus reducing the ecological hazards caused by the non-digestible components of the current antibiotics’ metabolites. Matzov et al 2017a, Auerbach-Nevo et al 2016
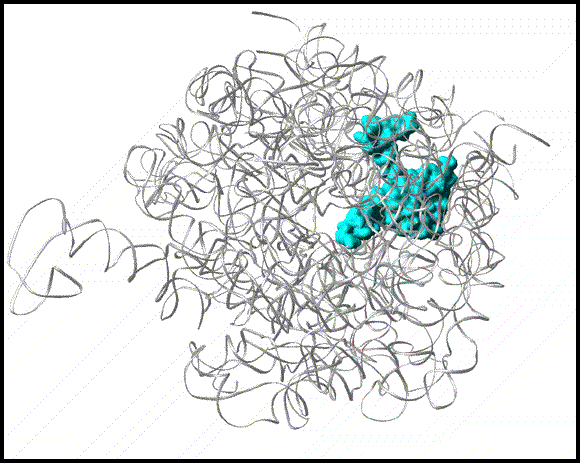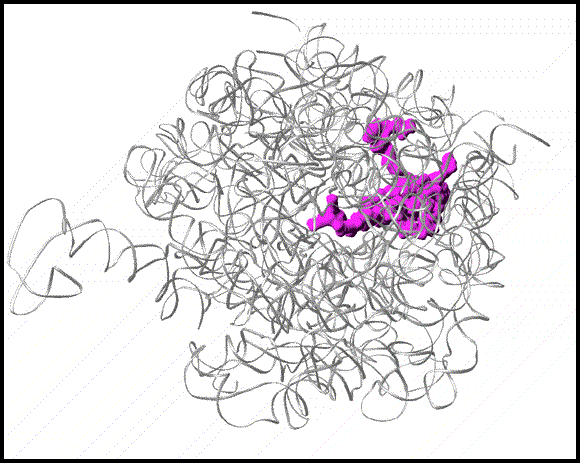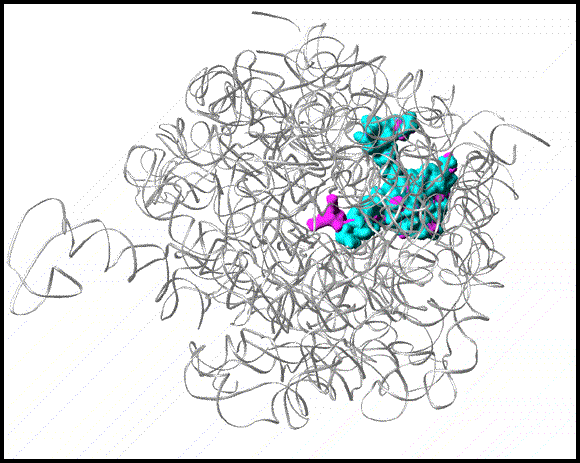
FIGURE 15:
Ribbon representation of the Staphylococcus aureus (SA) large ribosomal subunit (SA50S) in grey. A few target sites, colored in cyan, are shown. The magenta arc indicated a potential next-generation antibiotics binding site.
Acknowledgments:
These studies could not be performed without the cooperation, assistance and advice of the staff of the synchrotron radiation facilities at EMBL and MPG at DESY, ID14 at EMBL/ESRF, and ID19/APS/ANL. The Max-Planck Society, the U.S. National Institutes of Health (GM34360), the German Ministry for Science and Technology (grant 05-641EA), and the Kimmelman Center for Macromolecular Assembly at the Weizmann Institute provided support.
Weizmann Institute of Science, Rehovot, 76100 Israel
Tel: (972) 8 934 3028/2541 Fax: (972) 8 934 4154/4136;
Email: ada.yonath@weizmann.ac.il