






Tumor-antigen-independent targeting of solid tumors by armored macrophage-directed anti-TREM2 CAR T cells
Read more about Publication category: Cancer Immunotherapy
Publication category: Cancer Immunotherapy
Macrophage-targeted immunocytokine leverages myeloid, T, and NK cell synergy for cancer immunotherapy
Read more about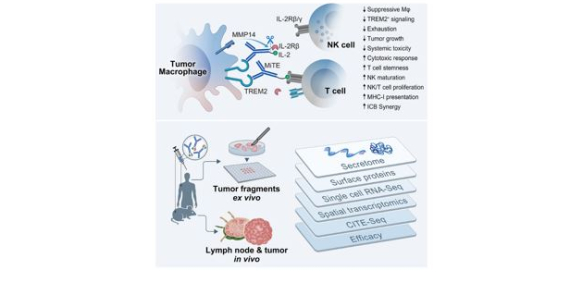 Publication category: Cancer Immunotherapy
Publication category: Cancer Immunotherapy
Distinct Cellular and Molecular Patterns in Pretreatment Peripheral Blood Are Associated with CAR T-cell Outcomes in Diffuse Large B-cell Lymphoma
Read more about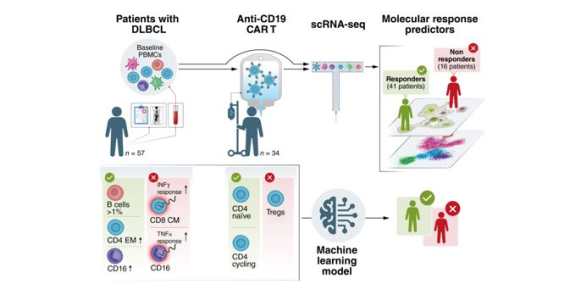 Publication category: Cancer Immunotherapy
Publication category: Cancer Immunotherapy
