Gideon Schreiber Lab

Research
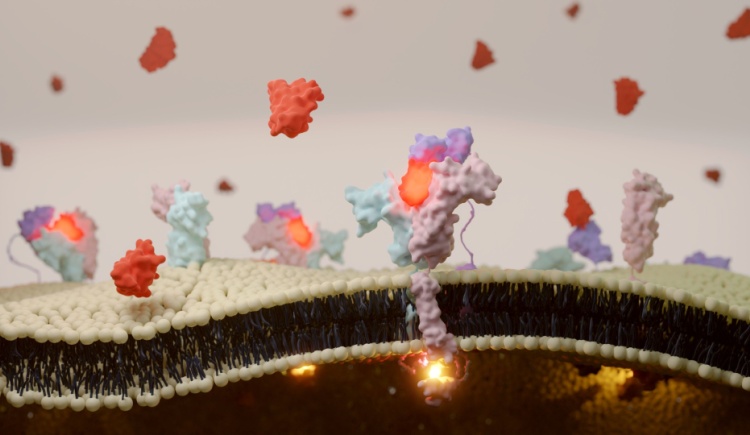
My laboratory is engaged in understanding the intricate web of protein-protein interactions that underpin every aspect of living organisms. With a sharp focus on the structural and functional nuances of these interactions, our research unravels the molecular dialogue that governs biological signaling. We employ a multifaceted, interdisciplinary strategy that seamlessly integrates protein engineering and bioinformatics. These tools act as pivotal inputs to address complex biological queries. Among our key research themes is a comprehensive elucidation of the interferon signaling pathway—a crucial component of immune response. Moreover, we engaged in tracking the evolutionary journey of the SARS-CoV-2 virus, providing vital insights into pandemic progression and viral behavior. Our most recent endeavor involves the pioneering design of specialized exosomes. These are engineered with precision to transport RNA molecules, opening new vistas in the realm of therapeutic delivery systems. Each discovery propels us closer to decoding the language of life's molecular machinery, which brings us closer to treat diseases in a specific manner.