Introducing rational changes in protein structure and regulatory sequences and visualizing the consequences of those changes in cells is a powerful way to characterize proteins' and cells' functions. This idea led us to collaborate with two laboratories (Maya Schuldiner, Weizmann Institute & Michael Knop, Heidelberg University) to create yeast “SWAT” libraries, which enable editing the N- or C- termini of all yeast genes in a customizable and massively parallel manner (Meurer et al. Nature Methods. 2018; Weill et al. Nature Methods. 2018). These libraries make it possible to create a new yeast library (e.g., all ORFs tagged with a sequence of interest) in one month, with minimal effort thanks to our robotic setup shown below.

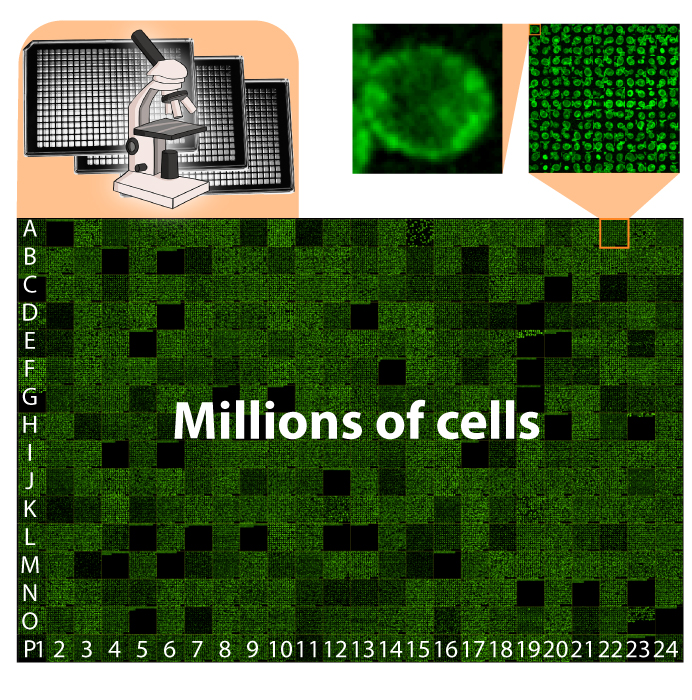
Working with yeast collections involves manipulating high-density arrays of printed cells, as well as acquiring and analyzing terabyte-sized image datasets. These data volumes led us to develop methods to manipulate cell arrays robotically, to image them by fluorescence microscopy, and to extract proteome information from the images. For example, we developed www.yeastRGB.org, (Dubreuil et al. Nuc. Acid. Res. 2019), which allows displaying, on a computer screen, the data of images that would cover a tennis court in their raw format. The picture below shows a montage of cells imaged in a 384-well glass-bottom plate.