Molecular analysis of DNA damage tolerance and mutagenesis in mammals
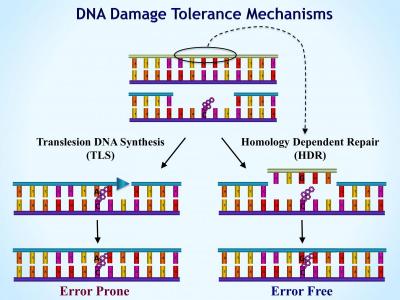 Despite the existence of multiple mechanisms that remove lesions from DNA, repair is usually not fully efficient, and leaves in the DNA unrepaired lesions. These lesions can interrupt DNA replication, leading to the formation of collapsed forks and single-stranded gaps, which if left untreated prevent completion of replication and can cause highly deleterious double strand breaks (DSB). DNA damage tolerance converts these structures to double-stranded DNA, without removing the lesion, by at least two mechanisms: Translesion DNA synthesis (TLS; also termed lesion bypass, or error-prone repair), or by homology-dependent repair (HDR; also termed homologous recombination repair, template switch or post-replication repair).
Despite the existence of multiple mechanisms that remove lesions from DNA, repair is usually not fully efficient, and leaves in the DNA unrepaired lesions. These lesions can interrupt DNA replication, leading to the formation of collapsed forks and single-stranded gaps, which if left untreated prevent completion of replication and can cause highly deleterious double strand breaks (DSB). DNA damage tolerance converts these structures to double-stranded DNA, without removing the lesion, by at least two mechanisms: Translesion DNA synthesis (TLS; also termed lesion bypass, or error-prone repair), or by homology-dependent repair (HDR; also termed homologous recombination repair, template switch or post-replication repair).
The key players in TLS are specialized DNA polymerases, which were discovered in our lab and in three additional labs. These TLS DNA polymerases are characterized by low fidelity, and the ability to carry out synthesis opposite lesions in DNA. Due to the miscoding nature of most DNA lesions, TLS is inherently mutagenic. In mammals TLS functions as a protective mechanism against cancer, as indicated by the high predisposition to cancer in the hereditary disease xeroderma pigmentosum variant, caused by germ line mutations in the TLS DNA polymerase . We are studying the molecular basis of the mechanism and regulation of TLS in mammalian cells, in order to elucidate its principles of operation, and its potential application for cancer prevention and therapy.