Zheng P., Furth N., O'Leary S. E.
, Shema E., Wanunu M. et al.
(2026)
Nano Letters.
26,
12,
p. 4143-4151
Despite advances in deciphering chromatin structure and dynamics in recent years, mapping the sequence position and combinatorial modifications of individual nucleosomes, the building blocks of chromatin, has yet to be achieved. In this work, we develop SM-NucSeq: a technology that combines single-molecule immunoaffinity detection with single-molecule real-time (SMRT) sequencing in zero-mode waveguides (ZMWs), which are sub-wavelength well-like structures that confine the depth of the excitation beam to the nanometer scale. We show that SM-NucSeq can detect histone modifications on intact nucleosomes and identify their underlying DNA sequences. We leverage the ZMW chips to load nucleosome/polymerase complexes and detect co-occurring histone modifications with fluorescent antibodies, validating each step on a chromatically resolved micromirror TIRF microscope. Finally, the application of SM-NucSeq reveals that DNA synthesis rates are reduced in nucleosomes compared to the double-stranded DNA control sample and that DNA synthesis rate profiles are histone-modification-dependent.
Aylon Y., Furth N., Pirona A. C.
, Lavie A., Fedorova O., Hassin O., Padrão N., Steinmetz M., Sarusi-Potuguez A., Fellus-Alyagor L., Shimoni I., Dassa B., Zwart W., Shema E., Oren M. et al.
(2025)
Proceedings of the National Academy of Sciences - PNAS.
122,
44,
e252264612.
Breast cancer is the leading cause of death in women under 50. The majority of breast cancers are estrogen receptor α-positive (ER+) and are commonly treated with hormonal therapies such as tamoxifen that inhibit ER activity. The TP53 tumor suppressor gene, encoding the p53 protein, is the most frequently mutated gene in breast cancer, and TP53 mutations are associated with diminished tamoxifen response and worse prognosis for breast cancer patients. Here, we report that in breast cancer cells p53 and ER cooperate to regulate the transcription of a set of genes encoding chromatin modifiers. The net result is a global increase in H3K4me3 and decrease in H3K9me3 chromatin marks. The resultant \u201copen\u201d chromatin is associated with increased transcription of luminal cell identity genes and enhanced tamoxifen sensitivity. Conversely, diminished p53 control of these chromatin modulators is associated with the evolution of tamoxifen resistance and cancer stem cell properties.
Griess O., Furth N., Harpaz N.
, Di Bernardo N., Salame T. M., Dassa B., Karagiannidis I., Isshiki Y., Gross M., Melnick A. M., Béguelin W., Ron G., Shema E. et al.
(2025)
PLoS Biology.
23,
6 ,
e3003191.
Diffuse large B cell lymphomas and follicular lymphomas show recurrent mutations in epigenetic regulators; among these are loss-of-function mutations in KMT2D and gain-of-function mutations in EZH2. To systematically explore the effects of these mutations on the wiring of the epigenetic network, we applied a single-cell approach to probe a wide array of histone modifications. We show that mutant-EZH2 elicits extensive effects on the epigenome of lymphomas, beyond alterations to H3K27 methylations, and is epistatic over KMT2D mutations. Utilizing the single-cell data, we present computational methods to measure epigenetic heterogeneity. We identify an unexpected characteristic of mutant-EZH2, but not KMT2D, in increasing heterogeneity, shedding light on a novel oncogenic mechanism mediated by this mutation. Finally, we present tools to reconstruct known interactions within the epigenetic network, as well as reveal potential novel cross talk between various modifications, supported by functional perturbations. Our work highlights novel roles for mutantEZH2 in lymphomagenesis and establishes new concepts for measuring epigenetic heterogeneity and intra-chromatin connectivity in cancer cells.
Furth N., Cohen N., Spitzer A.
, Salame T. M., Dassa B., Mehlman T., Brandis A., Moussaieff A., Friedmann-Morvinski D., Castro M. G., Fortin J., Suvà M. L., Tirosh I., Erez A., Ron G., Shema E. et al.
(2025)
Proceedings of the National Academy of Sciences - PNAS.
122,
1,
e240386212.
Malignant gliomas are heterogeneous tumors, mostly incurable, arising in the central nervous system (CNS) driven by genetic, epigenetic, and metabolic aberrations. Mutations in isocitrate dehydrogenase (IDH1/2mut) enzymes are predominantly found in low-grade gliomas and secondary high-grade gliomas, with IDH1 mutations being more prevalent. Mutant-IDH1/2 confers a gain-of-function activity that favors the conversion of a-ketoglutarate (α-KG) to the oncometabolite 2-hydroxyglutarate (2-HG), resulting in an aberrant hypermethylation phenotype. Yet, the complete depiction of the epigenetic alterations in IDHmut cells has not been thoroughly explored. Here, we applied an unbiased approach, leveraging epigenetic-focused cytometry by time-of-flight (CyTOF) analysis, to systematically profile the effect of mutant-IDH1 expression on a broad panel of histone modifications at single-cell resolution. This analysis revealed extensive remodeling of chromatin patterns by mutant-IDH1, with the most prominent being deregulation of histone acetylation marks. The loss of histone acetylation occurs rapidly following mutant-IDH1 induction and affects acetylation patterns over enhancers and intergenic regions. Notably, the changes in acetylation are not predominantly driven by 2-HG, can be rescued by pharmacological inhibition of mutant-IDH1, and reversed by acetate supplementations. Furthermore, cells expressing mutant-IDH1 show higher epigenetic and transcriptional heterogeneity and upregulation of oncogenes such as KRAS and MYC, highlighting its tumorigenic potential. Our study underscores the tight interaction between chromatin and metabolism dysregulation in glioma and highlights epigenetic and oncogenic pathways affected by mutant-IDH1-driven metabolic rewiring.
Erez N., Furth N., Fedyuk V.
, Wadden J., Aittaleb R., Adam T., Schwark K., Niculcea M., Miclea M., Mody R., Franson A., Parmar H. A., Ibrahim M., Lau B., Eze A., Nourmohammadi N., Fried I., Nazarian J., Ron G., Venneti S., Koschmann C., Shema E. et al.
(2025)
Cell Reports Medicine.
6,
1,
101918.
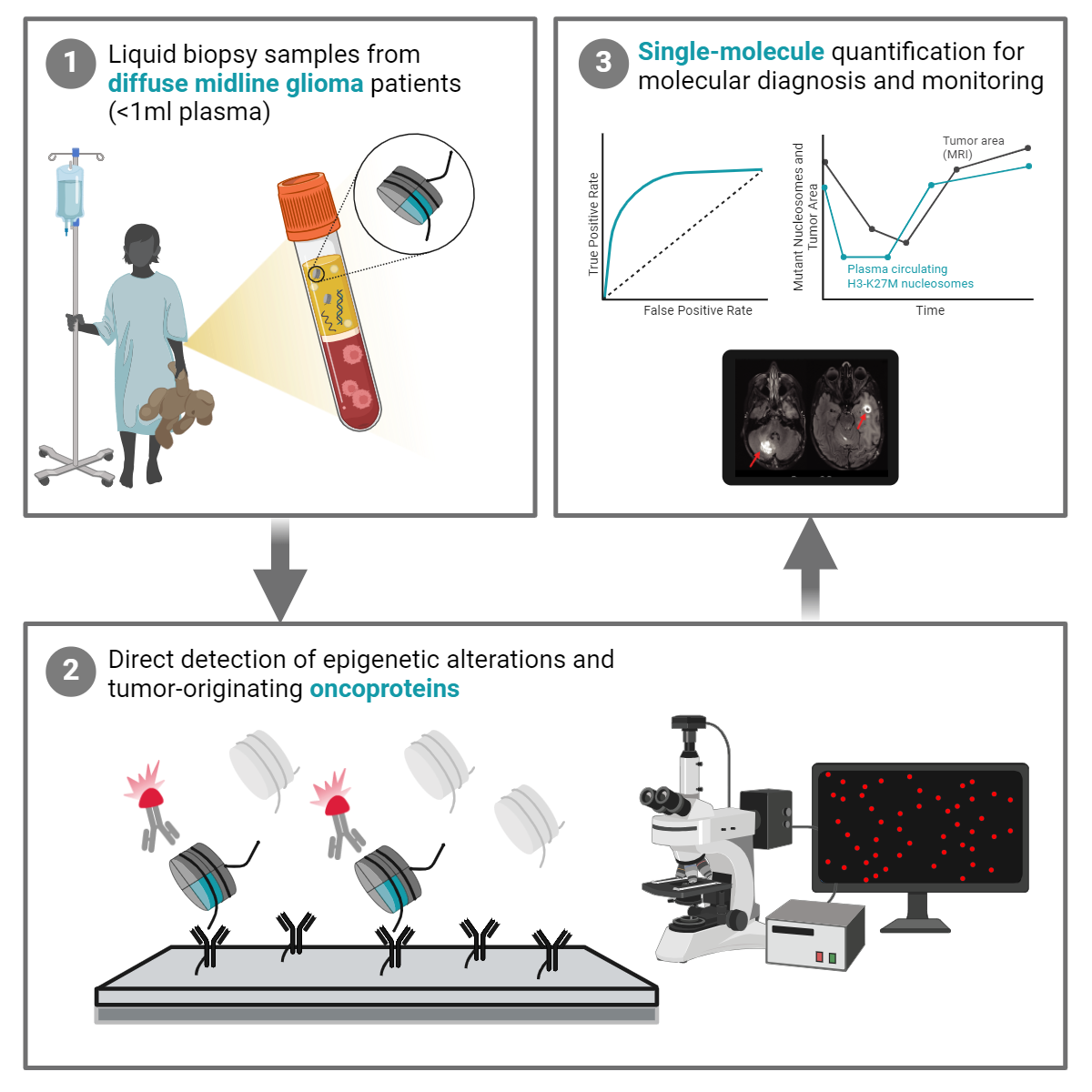
The analysis of cell-free tumor DNA (ctDNA) and proteins in the blood of patients with cancer potentiates a new generation of non-invasive diagnostic approaches. However, confident detection of tumor-originating markers is challenging, especially in the context of brain tumors, where these analytes in plasma are extremely scarce. Here, we apply a sensitive single-molecule technology to profile multiple histone modifications on individual nucleosomes from the plasma of patients with diffuse midline glioma (DMG). The system reveals epigenetic patterns unique to DMG, significantly differentiating this group of patients from healthy subjects or individuals diagnosed with other cancer types. We further develop a method to directly quantify the tumor-originating oncoproteins, lysine 27 to methionine substitution in histone H3 (H3-K27M) and mutant p53, from
Isshiki Y., Chen X., Teater M.
, Karagiannidis I., Nam H., Cai W., Meydan C., Xia M., Shen H., Gutierrez J., Easwar Kumar V., Carrasco S. E., Ouseph M. M., Yamshon S., Martin P., Griess O., Shema E., Porazzi P., Ruella M., Brentjens R. J., Inghirami G., Zappasodi R., Chadburn A., Melnick A. M., Béguelin W. et al.
(2025)
Cancer Cell.
43,
1,
p. 49-68.e9
T cell-based immunotherapies have demonstrated effectiveness in treating diffuse large B cell lymphoma (DLBCL) and follicular lymphoma (FL) but predicting response and understanding resistance remains a challenge. To address this, we developed syngeneic models reflecting the genetics, epigenetics, and immunology of human FL and DLBCL. We show that EZH2 inhibitors reprogram these models to re-express T cell engagement genes and render them highly immunogenic. EZH2 inhibitors do not harm tumor-controlling T cells or CAR-T cells. Instead, they reduce regulatory T cells, promote memory chimeric antigen receptor (CAR) CD8 phenotypes, and reduce exhaustion, resulting in a decreased tumor burden. Intravital 2-photon imaging shows increased CAR-T recruitment and interaction within the tumor microenvironment, improving lymphoma cell killing. Therefore, EZH2 inhibition enhances CAR-T cell efficacy through direct effects on CAR-T cells, in addition to rendering lymphoma B cells immunogenic. This approach is currently being evaluated in two clinical trials, NCT05934838 and NCT05994235, to improve immunotherapy outcomes in B cell lymphoma patients.
Algranati D., Oren R., Dassa B.
, Fellus-Alyagor L., Plotnikov A., Barr H., Harmelin A., London N., Ron G., Furth N., Shema E. et al.
(2024)
eLife.
13,
RP96257.
Diffuse midline gliomas (DMGs) are aggressive and fatal pediatric tumors of the central nervous system that are highly resistant to treatments. Lysine to methionine substitution of residue 27 on histone H3 (H3-K27M) is a driver mutation in DMGs, reshaping the epigenetic landscape of these cells to promote tumorigenesis. H3-K27M gliomas are characterized by deregulation of histone acetylation and methylation pathways, as well as the oncogenic MYC pathway. In search of effective treatment, we examined the therapeutic potential of dual targeting of histone deacetylases (HDACs) and MYC in these tumors. Treatment of H3-K27M patient- derived cells with Sulfopin, an inhibitor shown to block MYC-driven tumors in vivo, in combination with the HDAC inhibitor Vorinostat, resulted in substantial decrease in cell viability. Moreover, transcriptome and epigenome profiling revealed synergistic effect of this drug combination in downregulation of prominent oncogenic pathways such as mTOR. Finally, in vivo studies of patient-derived orthotopic xenograft models showed significant tumor growth reduction in mice treated with the drug combination. These results highlight the combined treatment with PIN1 and HDAC inhibitors as a promising therapeutic approach for these aggressive tumors.
Lim L. Q. J., Adler L., Hajaj E.
, Soria L. R., Perry R. B. T., Darzi N., Brody R., Furth N., Lichtenstein M., Bab-Dinitz E., Porat Z., Melman T., Brandis A., Aylon Y., Ben-Dor S., Orr I., Pri-Or A., Seger R., Shaul Y., Ruppin E., Oren M., Perez M., Meier J., Brunetti-Pierri N., Shema E., Ulitsky I., Erez A., Malitsky S., Itkin M. et al.
(2024)
Nature metabolism.
6,
7,
p. 1294-1309
Downregulation of the urea cycle enzyme argininosuccinate synthase (ASS1) in multiple tumors is associated with a poor prognosis partly because of the metabolic diversion of cytosolic aspartate for pyrimidine synthesis, supporting proliferation and mutagenesis owing to nucleotide imbalance. Here, we find that prolonged loss of ASS1 promotes DNA damage in colon cancer cells and fibroblasts from subjects with citrullinemia type I. Following acute induction of DNA damage with doxorubicin, ASS1 expression is elevated in the cytosol and the nucleus with at least a partial dependency on p53; ASS1 metabolically restrains cell cycle progression in the cytosol by restricting nucleotide synthesis. In the nucleus, ASS1 and ASL generate fumarate for the succination of SMARCC1, destabilizing the chromatin-remodeling complex SMARCC1SNF5 to decrease gene transcription, specifically in a subset of the p53-regulated cell cycle genes. Thus, following DNA damage, ASS1 is part of the p53 network that pauses cell cycle progression, enabling genome maintenance and survival. Loss of ASS1 contributes to DNA damage and promotes cell cycle progression, likely contributing to cancer mutagenesis and, hence, adaptability potential.
Alcolea M. P., Alonso-Curbelo D., Ambrogio C.
, Bullman S., Correia A. L., Ernst A., Halbrook C. J., Kelly G. L., Lund A. W., Quail D. F., Ruscetti M., Shema E., Stromnes I. M., Tam W. L. et al.
(2024)
Cancer Discovery.
14,
4,
p. 674-682
Shema E.
(2023)
Nature Reviews Cancer.
23,
5,
p. 271
In this article, Efrat Shema describes EPINUC, a liquid biopsy method based on epigenetic profiling of nucleosomes from cell-free DNA in the plasma. EPINUC, combined with protein biomarker measurements, enables the accurate differentiation of samples from healthy volunteers and patients.
Cohen L. R. Z., Kaffe B., Deri E.
, Leibson C., Nissim-Rafinia M., Maman M., Harpaz N., Ron G., Shema E., Meshorer E. et al.
(2023)
Nucleic Acids Research.
51,
4,
p. 1662-1673
The histone H3 variant, H3.3, is localized at specific regions in the genome, especially promoters and active enhancers, and has been shown to play important roles in development. A lysine to methionine substitution in position 27 (H3.3K27M) is a main cause of Diffuse Intrinsic Pontine Glioma (specifically Diffuse Midline Glioma, K27M-mutant), a lethal type of pediatric cancer. H3.3K27M has a dominant-negative effect by inhibiting the Polycomb Repressor Complex 2 (PRC2) activity. Here, we studied the immediate, genome-wide, consequences of the H3.3K27M mutation independent of PRC2 activity. We developed Doxycycline (Dox)-inducible mouse embryonic stem cells (ESCs) carrying a single extra copy of WT-H3.3, H3.3K27M and H3.3K27L, all fused to HA. We performed RNA-Seq and ChIP-Seq at different times following Dox induction in undifferentiated and differentiated ESCs. We find increased binding of H3.3 around transcription start sites in cells expressing both H3.3K27M and H3.3K27L compared with WT, but not in cells treated with PRC2 inhibitors. Differentiated cells carrying either H3.3K27M or H3.3K27L retain expression of ESC-active genes, in expense of expression of genes related to neuronal differentiation. Taken together, our data suggest that a modifiable H3.3K27 is required for proper histone incorporation and cellular maturation, independent of PRC2 activity.
Fedyuk V., Erez N., Furth N.
, Beresh O., Andreishcheva E. N., Shinde A., Jones D., Bar Zakai B., Mavor Y., Peretz T., Hubert A., Cohen J., Salah A., Temper M., Grinshpun A., Maoz M., Zick A., Ron G., Shema E. et al.
(2023)
Nature Biotechnology.
41,
2,
p. 212-221
The analysis of cell-free DNA (cfDNA) in plasma provides information on pathological processes in the body. Blood cfDNA is in the form of nucleosomes, which maintain their tissue- and cancer-specific epigenetic state. We developed a single-molecule multiparametric assay to comprehensively profile the epigenetics of plasma-isolated nucleosomes (EPINUC), DNA methylation and cancer-specific protein biomarkers. Our system allows for high-resolution detection of six active and repressive histone modifications and their ratios and combinatorial patterns on millions of individual nucleosomes by single-molecule imaging. In addition, our system provides sensitive and quantitative data on plasma proteins, including detection of non-secreted tumor-specific proteins, such as mutant p53. EPINUC analysis of a cohort of 63 colorectal cancer, 10 pancreatic cancer and 33 healthy plasma samples detected cancer with high accuracy and sensitivity, even at early stages. Finally, combining EPINUC with direct single-molecule DNA sequencing revealed the tissue of origin of colorectal, pancreatic, lung and breast tumors. EPINUC provides multilayered information of potential clinical relevance from limited (
Aylon Y., Furth N., Mallel G.
, Friedlander G., Nataraj N. B., Dong M., Hassin O., Zoabi R., Cohen B., Drendel V., Salame T. M., Mukherjee S., Harpaz N., Johnson R., Aulitzky W. E., Yarden Y., Shema E., Oren M. et al.
(2023)
Nature Communications.
14,
1,
133.
In this article the acknowledgements of Efrat Shema are omitted and should have read E.S is supported by the Emerson Collective and the Israel Cancer Research Fund, and is an incumbent of the Lisa and Jeffrey Aronin Family Career Development chair. The original article has been corrected.
Aylon Y., Furth N., Mallel G.
, Friedlander G., Nataraj N. B., Dong M., Hassin O., Zoabi R., Cohen B., Drendel V., Salame T. M., Mukherjee S., Harpaz N., Johnson R., Aulitzky W. E., Yarden Y., Shema E., Oren M. et al.
(2022)
Nature Communications.
13,
1,
7199.
Breast cancer, the most frequent cancer in women, is generally classified into several distinct histological and molecular subtypes. However, single-cell technologies have revealed remarkable cellular and functional heterogeneity across subtypes and even within individual breast tumors. Much of this heterogeneity is attributable to dynamic alterations in the epigenetic landscape of the cancer cells, which promote phenotypic plasticity. Such plasticity, including transition from luminal to basal-like cell identity, can promote disease aggressiveness. We now report that the tumor suppressor LATS1, whose expression is often downregulated in human breast cancer, helps maintain luminal breast cancer cell identity by reducing the chromatin accessibility of genes that are characteristic of a \u201cbasal-like\u201d state, preventing their spurious activation. This is achieved via interaction of LATS1 with the NCOR1 nuclear corepressor and recruitment of HDAC1, driving histone H3K27 deacetylation near NCOR1-repressed \u201cbasal-like\u201d genes. Consequently, decreased expression of LATS1 elevates the expression of such genes and facilitates slippage towards a more basal-like phenotypic identity. We propose that by enforcing rigorous silencing of repressed genes, the LATS1-NCOR1 axis maintains luminal cell identity and restricts breast cancer progression.
Harpaz N., Mittelman T., Beresh O.
, Griess O., Furth N., Salame T., Oren R., Fellus-Alyagor L., Harmelin A., Alexandrescu S., Marques J. G., Filbin M. G., Ron G., Shema E. et al.
(2022)
Molecular Cell.
82,
14,
p. 2696-2713+
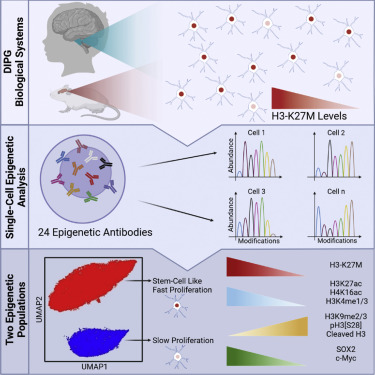
Cancer cells are highly heterogeneous at the transcriptional level and epigenetic state. Methods to study epigenetic heterogeneity are limited in throughput and information obtained per cell. Here, we adapted cytometry by time-of-flight (CyTOF) to analyze a wide panel of histone modifications in primary tumor-derived lines of diffused intrinsic pontine glioma (DIPG). DIPG is a lethal glioma, driven by a histone H3 lysine 27 mutation (H3-K27M). We identified two epigenetically distinct subpopulations in DIPG, reflecting inherent heterogeneity in expression of the mutant histone. These two subpopulations are robust across tumor lines derived from different patients and show differential proliferation capacity and expression of stem cell and differentiation markers. Moreover, we demonstrate the use of these high-dimensional data to elucidate potential interactions between histone modifications and epigenetic alterations during the cell cycle. Our work establishes new concepts for the analysis of epigenetic heterogeneity in cancer that could be applied to diverse biological systems.
Furth N., Algranati D., Dassa B.
, Beresh O., Fedyuk V., Morris N., Kasper L. H., Jones D., Monje M., Baker S. J., Shema E. et al.
(2022)
Cell Reports.
39,
7,
110836.
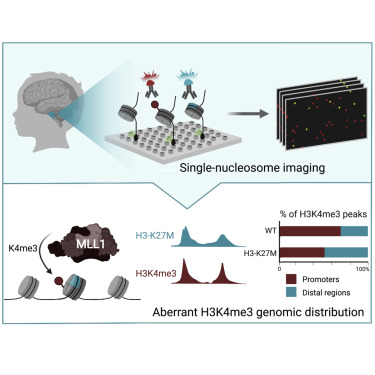
Cancer-associated mutations in genes encoding histones dramatically reshape chromatin and support tumorigenesis. Lysine to methionine substitution of residue 27 on histone H3 (K27M) is a driver mutation in high-grade pediatric gliomas, known to abrogate polycomb repressive complex 2 (PRC2) activity. We applied single-molecule systems to image individual nucleosomes and delineate the combinatorial epigenetic patterns associated with H3-K27M expression. We found that chromatin marks on H3-K27M-mutant nucleosomes are dictated both by their incorporation preferences and by intrinsic properties of the mutation. Mutant nucleosomes not only preferentially bind PRC2 but also directly interact with MLL1, leading to genome-wide redistribution of H3K4me3. H3-K27M-mediated deregulation of repressive and active chromatin marks leads to unbalanced \u201cbivalent\u201d chromatin, which may support a poorly differentiated cellular state. This study provides evidence for a direct effect of H3-K27M oncohistone on the MLL1-H3K4me3 pathway and highlights the capability of single-molecule tools to reveal mechanisms of chromatin deregulation in cancer.
Furth N. & Shema E.
(2022)
Current Opinion in Genetics and Development.
73,
101899.
Genome regulation is governed by the dynamics of chromatin modifications. The extensive and diverse array of DNA and histone modifications allow multiple elements to act combinatorically and direct tissue-specific and cell-specific outcomes. Yet, our ability to elucidate these complex combinations and link them to normal genome regulation, as well as understand their deregulation in cancer, has been hindered by the lack of suitable technologies. Here, we describe recent findings indicating the importance of the combinatorial epigenome, and novel methodologies to measure and characterize these combinations. These complementary methods span multiple disciplines, providing a means to decode epigenetic combinations and link them to biological outcomes. Finally, we discuss the promise of harnessing the rich combinatorial epigenetic information to improve cancer diagnostics and monitoring.
Sheban D., Shani T., Maor R.
, Aguilera-Castrejon A., Mor N., Oldak B., Shmueli M. D., Eisenberg-Lerner A., Bayerl J., Hebert J., Viukov S., Chen G., Kacen A., Krupalnik V., Chugaeva V., Tarazi S., Rodríguez-delaRosa A., Zerbib M., Ulman A., Masarwi S., Kupervaser M., Levin Y., Shema E., David Y., Novershtern N., Hanna J. H., Merbl Y. et al.
(2022)
Molecular Cell.
82,
1,
p. 106-122.e9
The fidelity of the early embryonic program is underlined by tight regulation of the chromatin. Yet, how the chromatin is organized to prohibit the reversal of the developmental program remains unclear. Specifically, the totipotency-to-pluripotency transition marks one of the most dramatic events to the chromatin, and yet, the nature of histone alterations underlying this process is incompletely characterized. Here, we show that linker histone H1 is post-translationally modulated by SUMO2/3, which facilitates its fixation onto ultra-condensed heterochromatin in embryonic stem cells (ESCs). Upon SUMOylation depletion, the chromatin becomes de-compacted and H1 is evicted, leading to totipotency reactivation. Furthermore, we show that H1 and SUMO2/3 jointly mediate the repression of totipotent elements. Lastly, we demonstrate that preventing SUMOylation on H1 abrogates its ability to repress the totipotency program in ESCs. Collectively, our findings unravel a critical role for SUMOylation of H1 in facilitating chromatin repression and desolation of the totipotent identity.
Kim K. L., van Galen P., Hovestadt V.
, Rahme G. J., Andreishcheva E. N., Shinde A., Gaskell E., Jones D. R., Shema E., Bernstein B. E. et al.
(2021)
Cell Reports Methods.
1,
5,
100061.
Epigenetic modifications control the stability and translation of mRNA molecules. Here, we present a microscopy-based platform for quantifying modified RNA molecules and for relating the modification patterns to single-cell phenotypes. We directly capture mRNAs from cell lysates on oligo-dT-coated coverslips, then visually detect and sequence individual m6A-immunolabled transcripts without amplification. Integration of a nanoscale device enabled us to isolate single cells on the platform, and thereby relate single-cell m6A modification states to gene expression signatures and cell surface markers. Application of the platform to MUTZ3 leukemia cells revealed a marked reduction in cellular m6A levels as CD34+ leukemic progenitors differentiate to CD14+ myeloid cells. We then coupled single-molecule m6A detection with fluorescence in situ hybridization (FISH) to relate mRNA and m6A levels of individual genes to single-cell phenotypes. This single-cell multi-modal assay suite can empower investigations of RNA modifications in rare populations and single cells.
Furth N., Shilo S., Cohen N.
, Erez N., Fedyuk V., Schrager A. M., Weinberger A., Dror A. A., Zigron A., Shehadeh M., Sela E., Srouji S., Amit S., Levy I., Segal E., Dahan R., Jones D., Douek D. C., Shema E. et al.
(2021)
PLoS ONE.
16,
7 July,
e0255096.
The COVID-19 pandemic raises the need for diverse diagnostic approaches to rapidly detect different stages of viral infection. The flexible and quantitative nature of single-molecule imaging technology renders it optimal for development of new diagnostic tools. Here we present a proof-of-concept for a single-molecule based, enzyme-free assay for detection of SARS-CoV-2. The unified platform we developed allows direct detection of the viral genetic material from patients' samples, as well as their immune response consisting of IgG and IgM antibodies. Thus, it establishes a platform for diagnostics of COVID-19, which could also be adjusted to diagnose additional pathogens.
Most (if not all) tumors emerge and progress under a strong evolutionary pressure imposed by trophic, metabolic, immunological, and therapeutic factors. The relative impact of these factors on tumor evolution changes over space and time, ultimately favoring the establishment of a neoplastic microenvironment that exhibits considerable genetic, phenotypic, and behavioral heterogeneity in all its components. Here, we discuss the main sources of intratumoral heterogeneity and its impact on the natural history of the disease, including sensitivity to treatment, as we delineate potential strategies to target such a detrimental feature of aggressive malignancies.
Shema E., Bernstein B. E. & Buenrostro J. D.
(2019)
Nature Genetics.
51,
1,
p. 19-25
Recent advances in single-cell and single-molecule epigenomic technologies now enable the study of genome regulation and dynamics at unprecedented resolution. In this Perspective, we highlight some of these transformative technologies and discuss how they have been used to identify new modes of gene regulation. We also contrast these assays with recent advances in single-cell transcriptomics and argue for the essential role of epigenomic technologies in both understanding cellular diversity and discovering gene regulatory mechanisms. In addition, we provide our view on the next generation of biological tools that we expect will open new avenues for elucidating the fundamental principles of gene regulation. Overall, this Perspective motivates the use of these high-resolution epigenomic technologies for mapping cell states and understanding regulatory diversity at single-molecule resolution within single cells.
Shema E., Jones D., Shoresh N.
, Donohue L., Ram O., Bernstein B. E. et al.
(2016)
Science.
352,
6286,
p. 717-721
Different combinations of histone modifications have been proposed to signal distinct gene regulatory functions, but this area is poorly addressed by existing technologies. We applied high-throughput single-molecule imaging to decode combinatorial modifications on millions of individual nucleosomes from pluripotent stem cells and lineage-committed cells.We identified definitively bivalent nucleosomes with concomitant repressive and activating marks, as well as other combinatorial modification states whose prevalence varies with developmental potency. We showed that genetic and chemical perturbations of chromatin enzymes preferentially affect nucleosomes harboring specific modification states. Last, we combined this proteomic platform with single-molecule DNA sequencing technology to simultaneously determine the modification states and genomic positions of individual nucleosomes. This single-molecule technology has the potential to address fundamental questions in chromatin biology and epigenetic regulation.
Tarcic O., Pateras I. S., Cooks T.
, Shema E., Kanterman J., Ashkenazi H., Boocholez H., Hubert A., Rotkopf R., Baniyash M., Pikarsky E., Gorgoulis V. G., Oren M. et al.
(2016)
Cell Reports.
14,
6,
p. 1462-1476
Factors linking inflammation and cancer are of great interest. We now report that the chromatin-targeting E3 ubiquitin ligase RNF20/RNF40, driving histone H2B monoubiquitylation (H2Bub1), modulates inflammation and inflammation-associated cancer in mice and humans. Downregulation of RNF20 and H2Bub1 favors recruitment of p65-containing nuclear factor κB (NF-κB) dimers over repressive p50 homodimers and decreases the heterochromatin mark H3K9me3 on a subset of NF-κB target genes to augment their transcription. Concordantly, RNF20+/- mice are predisposed to acute and chronic colonic inflammation and inflammation-associated colorectal cancer, with excessive myeloid-derived suppressor cells (MDSCs) that may quench antitumoral T cell activity. Notably, colons of human ulcerative colitis patients, as well as colorectal tumors, reveal downregulation of RNF20/RNF40 and H2Bub1 in both epithelium and stroma, supporting the clinical relevance of our tissue culture and mouse model findings.
Shema E., Nikolov M., Haj-Yahya M.
, Siman P., Allemand E., Yamaguchi Y., Muchardt C., Urlaub H., Brik A., Oren M., Fischle W. et al.
(2013)
Cell Reports.
4,
3,
p. 601-608
Chromatin posttranslational modifications (PTMs), including monoubiquitylation of histone H2B on lysine 120 (H2Bub1), play a major role in regulating genome functions. To elucidate the molecular mechanisms of H2Bub1 activity, a chromatin template uniformly containing H2Bub1 was used as an affinity matrix to identify preferentially interacting human proteins. Over 90 such factors were found, including proteins and protein complexes associated with transcription, RNA posttranscriptional modifications, and DNA replication and repair. Notably, we found that the SWI/SNF chromatin remodeling complex associates preferentially with H2Bub1-rich chromatin. Moreover,SWI/SNF is required for optimal transcription of a subset of genes that are selectively dependent on H2Bub1. Our findings substantially expand the known H2Bub1 interactome and provide insights into the functions of this PTM in mammalian gene regulation
Haj-Yahya M., Eltarteer N., Ohayon S.
, Shema E., Kotler E., Oren M., Brik A. et al.
(2012)
ANGEWANDTE CHEMIE-INTERNATIONAL EDITION.
51,
46,
p. 11535-11539
Stable like a rock: An efficient method to generate N-methylated isopeptide bonds has been developed. The strategy was used to generate highly stable ubiquitinated peptides and proteins that are resistant to deubiquitinases (DUBs; see scheme). Thus, the behavior of several stable ubiquitin conjugates with different DUBs was studied in-vitro and within a cellular environment.
Fuchs G., Shema E., Vesterman R.
, Kotler E., Wolchinsky Z., Wilder S., Golomb L., Pribluda A., Zhang F., Haj-Yahya M., Feldmesser E., Brik A., Yu X., Hanna J., Aberdam D., Domany E., Oren M. et al.
(2012)
Molecular Cell.
46,
5,
p. 662-673
Embryonic stem cells (ESCs) maintain high genomic plasticity, which is essential for their capacity to enter diverse differentiation pathways. Posttranscriptional modifications of chromatin histones play a pivotal role in maintaining this plasticity. We now report that one such modification, monoubiquitylation of histone H2B on lysine 120 (H2Bub1), catalyzed by the E3 ligase RNF20, increases during ESC differentiation and is required for efficient execution of this process. This increase is particularly important for the transcriptional induction of relatively long genes during ESC differentiation. Furthermore, we identify the deubiquitinase USP44 as a negative regulator of H2B ubiquitylation, whose downregulation during ESC differentiation contributes to the increase in H2Bub1. Our findings suggest that optimal ESC differentiation requires dynamic changes in H2B ubiquitylation patterns, which must occur in a timely and well-coordinated manner.
Shiloh Y., Shema E., Moyal L.
, Oren M. et al.
(2011)
FEBS Letters.
585,
18,
p. 2795-2802
The DNA damage response (DDR) is emerging as a vast signaling network that temporarily modulates numerous aspects of cellular metabolism in the face of DNA lesions, especially critical ones such as the double strand break (DSB). The DDR involves extensive dynamics of protein post-translational modifications, most notably phosphorylation and ubiquitylation. The DSB response is mobilized primarily by the ATM protein kinase, which phosphorylates a plethora of key players in its various branches. It is based on a core of proteins dedicated to the damage response, and a cadre of proteins borrowed temporarily from other cellular processes to help meet the challenge. A recently identified novel component of the DDR pathway - histone H2B monoubiquitylation - exemplifies this principle. In mammalian cells, H2B monoubiquitylation is driven primarily by an E3 ubiquitin ligase composed of the two RING finger proteins RNF20 and RNF40. Generation of monoubiquitylated histone H2B (H2Bub) has been known to be coupled to gene transcription, presumably modulating chromatin decondensation at transcribed regions. New evidence indicates that the regulatory function of H2Bub on gene expression can selectively enhance or suppress the expression of distinct subsets of genes through a mechanism involving the hPAF1 complex and the TFIIS protein. This delicate regulatory process specifically affects genes that control cell growth and genome stability, and places RNF20 and RNF40 in the realm of tumor suppressor proteins. In parallel, it was found that following DSB induction, the H2B monoubiquitylation module is recruited to damage sites where it induces local H2Bub, which in turn is required for timely recruitment of DSB repair protein and, subsequently, timely DSB repair. This pathway represents a crossroads of the DDR and chromatin organization, and is a typical example of how the DDR calls to action functional modules that in unprovoked cells regulate other processes.
Shema E., Oren M. & Minsky N.
(2011)
Methods.
54,
3,
p. 326-330
Histone H2B ubiquitylation was shown to be associated with actively transcribed genes in mammalian cells and has been suggested to be involved in transcriptional regulation. Despite the limited applicability of genetic tools to analyze H2B ubiquitylation in mammals, several biochemical and immunological approaches have been successfully implemented to study this modification. Here we describe several techniques to detect ubiquitylated H2B in mammalian cells and to dissect its genomic localization.
Dori-Bachash M., Shema E. & Tirosh I.
(2011)
PLoS Biology.
9,
7,
e1001106.
mRNA levels are determined by the balance between transcription and mRNA degradation, and while transcription has been extensively studied, very little is known regarding the regulation of mRNA degradation and its coordination with transcription. Here we examine the evolution of mRNA degradation rates between two closely related yeast species. Surprisingly, we find that around half of the evolutionary changes in mRNA degradation were coupled to transcriptional changes that exert opposite effects on mRNA levels. Analysis of mRNA degradation rates in an interspecific hybrid further suggests that opposite evolutionary changes in transcription and in mRNA degradation are mechanistically coupled and were generated by the same individual mutations. Coupled changes are associated with divergence of two complexes that were previously implicated both in transcription and in mRNA degradation (Rpb4/7 and Ccr4-Not), as well as with sequence divergence of transcription factor binding motifs. These results suggest that an opposite coupling between the regulation of transcription and that of mRNA degradation has shaped the evolution of gene regulation in yeast.
Shema E., Kim J., Roeder R. G.
, Oren M. et al.
(2011)
Molecular Cell.
42,
4,
p. 477-488
hBRE1/RNF20 is the major E3 ubiquitin ligase for histone H2B. RNF20 depletion causes a global reduction of monoubiquitylated H2B (H2Bub) levels and augments the expression of growth-promoting, pro-oncogenic genes. Those genes reside preferentially in compact chromatin and are inefficiently transcribed under basal conditions. We now report that RNF20, presumably via H2Bub, selectively represses those genes by interfering with chromatin recruitment of TFIIS, a factor capable of relieving stalled RNA polymerase II. RNF20 inhibits the interaction between TFIIS and the PAF1 complex and hinders transcriptional elongation. TFIIS ablation selectively abolishes the upregulation of those genes upon RNF20 depletion and attenuates the cellular response to EGF. Consistent with its positive role in transcription of pro-oncogenic genes, TFIIS expression is elevated in various human tumors. Our findings provide a molecular mechanism for selective gene repression by RNF20 and position TFIIS as a key target of RNF20's tumor suppressor activity.
Zwang Y., Sas-Chen A., Drier Y.
, Shay T., Avraham R., Lauriola M., Shema E., Lidor Nili N. E., Jacob-Hirsch J., Amariglio N., Lu Y., Mills G. B., Rechavi G., Oren M., Domany E., Yarden Y. et al.
(2011)
Molecular Cell.
42,
4,
p. 524-535
Normal cells require continuous exposure to growth factors in order to cross a restriction point and commit to cell-cycle progression. This can be replaced by two short, appropriately spaced pulses of growth factors, where the first pulse primes a process, which is completed by the second pulse, and enables restriction point crossing. Through integration of comprehensive proteomic and transcriptomic analyses of each pulse, we identified three processes that regulate restriction point crossing: (1) The first pulse induces essential metabolic enzymes and activates p53-dependent restraining processes. (2) The second pulse eliminates, via the PI3K/AKT pathway, the suppressive action of p53, as well as (3) sets an ERK-EGR1 threshold mechanism, which digitizes graded external signals into an all-or-none decision obligatory for S phase entry. Together, our findings uncover two gating mechanisms, which ensure that cells ignore fortuitous growth factors and undergo proliferation only in response to consistent mitogenic signals.
Moyal L., Lerenthal Y., Gana-Weisz M.
, Mass G., So S., Wang S., Eppink B., Chung Y. M., Shalev G., Shema E., Shkedy D., Smorodinsky N. I., van Vliet V. N., Kuster B., Mann M., Ciechanover A., Dahm-Daphi J., Kanaar R., Hu M. C. -. et al.
(2011)
Molecular Cell.
41,
5,
p. 529-542
The cellular response to DNA double-strand breaks (DSBs) is mobilized by the protein kinase ATM, which phosphorylates key players in the DNA damage response (DDR) network. A major question is how ATM controls DSB repair. Optimal repair requires chromatin relaxation at damaged sites. Chromatin reorganization is coupled to dynamic alterations in histone posttranslational modifications. Here, we show that in human cells, DSBs induce monoubiquitylation of histone H2B, a modification that is associated in undamaged cells with transcription elongation. We find that this process relies on recruitment to DSB sites and ATM-dependent phosphorylation of the responsible E3 ubiquitin ligase: the RNF20-RNF40 heterodimer. H2B monoubiquitylation is required for timely recruitment of players in the two major DSB repair pathways-nonhomologous end-joining and homologous recombination repair-and optimal repair via both pathways. Our data and previous data suggest a two-stage model for chromatin decondensation that facilitates DSB repair.
Pirngruber J., Shchebet A., Schreiber L.
, Shema E., Minsky N., Chapman R. D., Eick D., Aylon Y., Oren M., Johnsen S. A. et al.
(2009)
EMBO Reports.
10,
8,
p. 894-900
Post-translational histone modifications have essential roles in controlling nuclear processes; however, the specific mechanisms regulating these modifications and their combinatorial activities remain elusive. Cyclin-dependent kinase 9 (CDK9) regulates gene expression by phosphorylating transcriptional regulatory proteins, including the RNA polymerase II carboxy-terminal domain. Here, we show that CDK9 activity is essential for maintaining global and gene-associated levels of histone H2B monoubiquitination (H2Bub1). Furthermore, CDK9 activity and H2Bub1 help to maintain correct replication-dependent histone messenger RNA (mRNA) 3-end processing. CDK9 knockdown consistently resulted in inefficient recognition of the correct mRNA 3-end cleavage site and led to increased read-through of RNA polymerase II to an alternative downstream polyadenylation signal. Thus, CDK9 acts to integrate phosphorylation during transcription with chromatin modifications to control co-transcriptional histone mRNA processing.
Shema E., Tirosh I., Aylon Y.
, Huang J., Ye C., Moskovits N., Raver-Shapira N., Minsky N., Pirngruber J., Tarcic G., Hublarova P., Moyal L., Gana-Weisz M., Shiloh Y., Yarden Y., Johnsen S. A., Vojtesek B., Berger S. L., Oren M. et al.
(2008)
GENES & DEVELOPMENT.
22,
19,
p. 2664-2676
Histone monoubiquitylation is implicated in critical regulatory processes. We explored the roles of histone H2B ubiquitylation in human cells by reducing the expression of hBREl/RNF20, the major H2B-specific E3 ubiquitin ligase. While H2B ubiquitylation is broadly associated with transcribed genes, only a subset of genes was transcriptionally affected by RNF20 depletion and abrogation of H2B ubiquitylation. Gene expression dependent on RNF20 includes histones H2A and H2B and the p53 tumor suppressor. In contrast, RNF20 suppresses the expression of several proto-oncogenes, which reside preferentially in closed chromatin and are modestly transcribed despite bearing marks usually associated with high transcription rates. Remarkably, RNF20 depletion augmented the transcriptional effects of epidermal growth factor (EGF), increased cell migration, and elicited transformation and tumorigenesis. Furthermore, frequent RNF20 promoter hypermethylation was observed in tumors. RNF20 may thus be a putative tumor suppressor, acting through selective regulation of a distinct subset of genes.
Minsky N., Shema E., Field Y.
, Schuster M., Segal E., Oren M. et al.
(2008)
Nature Cell Biology.
10,
4,
p. 483-488
Histone modifications have emerged as important regulators of transcription. Histone H2B monoubiquitination has also been implicated in transcription; however, better understanding of the biological significance of this modification in mammalian cells has been hindered by the lack of suitable reagents, particularly antibodies capable of specifically recognizing ubiquitinated H2B (ubH2B). Here, we report the generation of anti-ubH2B monoclonal antibodies using a branched peptide as immunogen. These antibodies provide a powerful tool for exploring the biochemical functions of H2B monoubiquitination at both a genome-wide and gene-specific level. Application of these antibodies in high resolution chromatin immunoprecipitation (ChIP)-chip experiments in human cells, using tiling arrays, revealed preferential association of ubiquitinated H2B with the transcribed regions of highly expressed genes. Unlike dimethylated H3K4, ubH2B was not associated with distal promoter regions. Furthermore, experimental modulation of the transcriptional activity of the tumour suppressor p53 was accompanied by rapid changes in the H2B ubiquitination status of its p21 target gene, attesting to the dynamic nature of this process. It has recently been demonstrated that the apparent extent of gene expression often reflects elongation rather than initiation rates; thus, our findings suggest that H2B ubiquitination is intimately linked with global transcriptional elongation in mammalian cells.
Klutstein M., Shaked H., Sherman A.
, Avivi-Ragolsky N., Shema E., Zenvirth D., Levy A., Simchen G. et al.
(2008)
Genetics.
178,
4,
p. 2389-2397
The Saccharomyces cerevisiae RAD54 gene has critical roles in DNA double-strand break repair, homologous recombination, and gene targeting. Previous results show that the yeast gene enhances gene targeting when expressed in Arabidopsis thaliana. In this work we address the trans-species compatibility of Rad54 functions. We show that overexpression of yeast RAD54 in Arabidopsis enhances DNA damage resistance severalfold. Thus, the yeast gene is active in the Arabidopsis homologous-recombination repair system. Moreover, we have identified an A. thaliana ortholog of yeast RAD54, named AtRAD54. This gene, with close sequence similarity to RAD54, complements methylmethane sulfonate (MMS) sensitivity but not UV sensitivity or gene targeting defects of rad54D mutant yeast cells. Overexpression of AtRAD54 in Arabidopsis leads to enhanced resistance to DNA damage. This gene's assignment as a RAD54 ortholog is further supported by the interaction of AtRad54 with AtRad51 and the interactions between alien proteins (i.e., yeast Rad54 with AtRAD51 and yeast Rad51 with AtRad54) in a yeast two-hybrid experiment. These interactions hint at the molecular nature of this interkingdom complementation, although the stronger effect of the yeast Rad54 in plants than AtRad54 in yeast might be explained by an ability of the Rad54 protein to act alone, independently of its interaction with Rad51.