Shlush Lab
hematopoietic system

Research
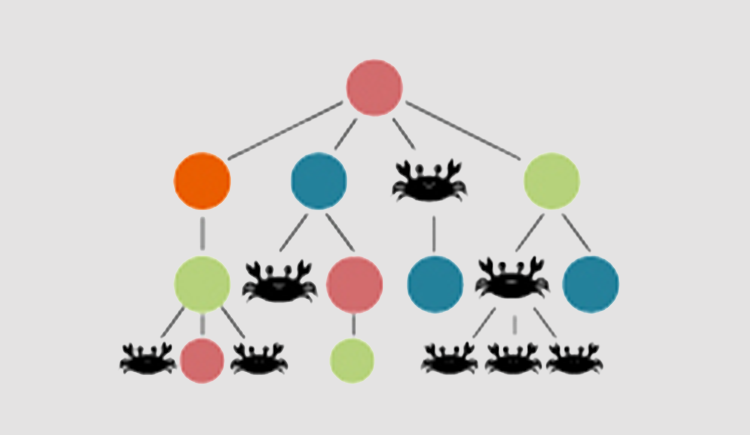
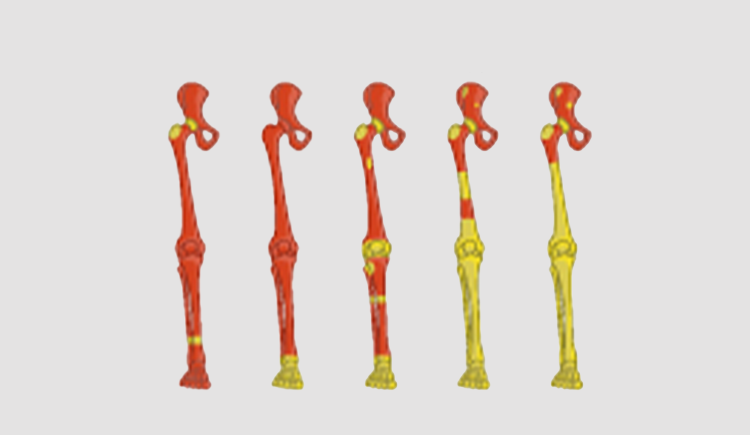
The aging of the human hematopoietic system is typically correlated with: 1) clonal hematopoiesis 2) myeloid malignancies 3) reduced T and B cell clonal diversity 4) reduced myeloid cells function 5) red to yellow marrow transition. Understanding the evolutionary principals driving these phenotypes is the first step in early diagnosis and treatment of hematopoiesis malfunction. Hematopoiesis failure in humans is a long process of somatic selection. Somatic evolution creates new clones exploring the fitness space. Without changes in the somatic environment new clones will loose to the young hematopoietic system which have been optimized over millions of years of germline evolution. Under changing environment, the fittest clones will gradually take over and in time due to antagonistic pleiotropy will lead to a disease. Our lab is a multidisciplinary lab composed of hematologists, evolutionary biologists and computationalsystem biologists all trying to understand the rules of human hematopoietic aging.