

Publications
2025
-
(2025) Cell Reports. 44, 12, 116583. Abstract
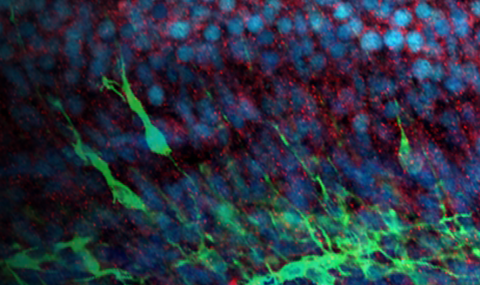
Neurological disorders are linked to mitochondrial dysfunction and calcium overload. Mitochondrial calcium uptake is mediated by the mitochondrial calcium uniporter (mtCU), regulated by MICU1, which can be either homodimerized or heterodimerized with MICU2 or MICU3. Though MICU2 is scarce in the adult brain, MICU2 loss in patients leads to a neurodevelopmental disorder. We hypothesized that MICU2 is required for developmental calcium signaling and neuronal migration. MICU2 is present in the developing mouse brain but disappears by maturation, contrasting with other mtCU subunits that increase. MICU2 loss in mice does not affect cytoplasmic calcium but augments the mitochondrial matrix calcium rise in primary cortical neurons, leading to neuronal overmigration in the cortex and behavioral changes at 2 but not 12 months. Consistently, mitochondrial calcium uptake is not significantly affected in the adult animal cortex. MICU2-deficient patient fibroblasts copy the mitochondria-confined calcium alteration in developing neurons. Thus, MICU2 is important during neurodevelopment, likely by regulating the mtCU, and is eliminated by brain maturation.
-
(2025) Nature Communications. 16, 1, 4094. Abstract
The viscoelastic properties of tissues influence their morphology and cellular behavior, yet little is known about changes in these properties during brain malformations. Lissencephaly, a severe cortical malformation caused by LIS1 mutations, results in a smooth cortex. Here, we show that human-derived brain organoids with LIS1 mutation exhibit increased stiffness compared to controls at multiple developmental stages. This stiffening correlates with abnormal extracellular matrix (ECM) expression and organization, as well as elevated water content, measured by diffusion-weighted MRI. Short-term MMP9 treatment reduces both stiffness and water diffusion levels to control values. Additionally, a computational microstructure mechanical model predicts mechanical changes based on ECM organization. These findings suggest that LIS1 plays a critical role in ECM regulation during brain development and that its mutation leads to significant viscoelastic alterations.
-
(2025) Nature. 639, 8054, p. 315-320 Abstract
As the field of neural organoids and assembloids expands, there is an emergent need for guidance and advice on designing, conducting and reporting experiments to increase the reproducibility and utility of these models. In this Perspective, we present a framework for the experimental process that encompasses ensuring the quality and integrity of human pluripotent stem cells, characterizing and manipulating neural cells in vitro, transplantation techniques and considerations for modelling human development, evolution and disease. As with all scientific endeavours, we advocate for rigorous experimental designs tailored to explicit scientific questions as well as transparent methodologies and data sharing to provide useful knowledge for current research practices and for developing regulatory standards.
-
(2025) Development (Cambridge). 152, 2, dev202703. Abstract
Proneural genes are conserved drivers of neurogenesis across the animal kingdom. How their functions have adapted to guide human-specific neurodevelopmental features is poorly understood. Here, we mined transcriptomic data from human fetal cortices and generated from human embryonic stem cell-derived cortical organoids (COs) to show that NEUROG1 and NEUROG2 are most highly expressed in basal neural progenitor cells, with pseudotime trajectory analyses indicating that NEUROG1-derived lineages predominate early and NEUROG2 lineages later. Using ChIP-qPCR, gene silencing and overexpression studies in COs, we show that NEUROG2 is necessary and sufficient to directly transactivate known target genes (NEUROD1, EOMES, RND2). To identify new targets, we engineered NEUROG2-mCherry knock-in human embryonic stem cells for CO generation. The mCherry-high CO cell transcriptome is enriched in extracellular matrix-associated genes, and two genes associated with human-accelerated regions: PPP1R17 and FZD8. We show that NEUROG2 binds COL1A1, COL3A1 and PPP1R17 regulatory elements, and induces their ectopic expression in COs, although NEUROG2 is not required for this expression. Neurog2 similarly induces Col3a1 and Ppp1r17 in murine P19 cells. These data are consistent with a conservation of NEUROG2 function across mammalian species.
-
(2025) ISBI 2025 - 2025 IEEE 22nd International Symposium on Biomedical Imaging, Proceedings. Abstract
Recent advances have enabled the study of human brain de-velopment using brain organoids derived from stem cells. Quantifying cellular processes like mitosis in these organoids offers insights into neurodevelopmental disorders, but the manual analysis is time-consuming, and existing datasets lack specific details for brain organoid studies. We introduce BOrg, a dataset designed to study mitotic events in the embryonic development of the brain using confocal microscopy images of brain organoids. BOrg utilizes an efficient annotation pipeline with sparse point annotations and techniques that minimize expert effort, overcoming limitations of standard deep learning approaches on sparse data. We adapt and benchmark state-of-the-art object detection and cell counting models on BOrg for detecting and analyzing mitotic cells across the prophase, metaphase, anaphase, and telophase stages. Our results demonstrate that these adapted models significantly improve mitosis analysis efficiency and accuracy for brain organoid research compared to existing methods. BOrg facilitates the development of automated tools to quantify statistics like mitosis rates, aiding mecha-nistic studies of neurodevelopmental processes and disorders. Data and code are available at BOrg's GitHub page.
2024
-
(2024) Journal of Clinical Investigation. 134, 21, e175435. Abstract
Brain size and cellular heterogeneity are tightly regulated by species-specific proliferation and differentiation of multipotent neural progenitor cells (NPCs). Errors in this process are among the mechanisms of primary hereditary microcephaly (MCPH), a group of disorders characterized by reduced brain size and intellectual disability. Biallelic citron rho-interacting serine/ threonine kinase (CIT) missense variants that disrupt kinase function (CITKI/KI) and frameshift loss-of-function variants (CITFS/FS) are the genetic basis for MCPH17; however, the function of CIT catalytic activity in brain development and NPC cytokinesis is unknown. Therefore, we created the CitKI/KImouse model and found that it did not phenocopy human microcephaly, unlike biallelic CitFS/FSanimals. Nevertheless, both Cit models exhibited binucleation, DNA damage, and apoptosis. To investigate human-specific mechanisms of CIT microcephaly, we generated CITKI/KIand CITFS/FShuman forebrain organoids. We found that CITKI/KIand CITFS/FSorganoids lost cytoarchitectural complexity, transitioning from pseudostratified to simple neuroepithelium. This change was associated with defects that disrupted the polarity of NPC cytokinesis, in addition to elevating apoptosis. Together, our results indicate that both CIT catalytic and scaffolding functions in NPC cytokinesis are critical for human corticogenesis. Species differences in corticogenesis and the dynamic 3D features of NPC mitosis underscore the utility of human forebrain organoid models for understanding human microcephaly.
-
(2024) Nature. 628, 8007, p. 391-399 Abstract
The human nervous system is a highly complex but organized organ. The foundation of its complexity and organization is laid down during regional patterning of the neural tube, the embryonic precursor to the human nervous system. Historically, studies of neural tube patterning have relied on animal models to uncover underlying principles. Recently, models of neurodevelopment based on human pluripotent stem cells, including neural organoids15 and bioengineered neural tube development models610, have emerged. However, such models fail to recapitulate neural patterning along both rostralcaudal and dorsalventral axes in a three-dimensional tubular geometry, a hallmark of neural tube development. Here we report a human pluripotent stem cell-based, microfluidic neural tube-like structure, the development of which recapitulates several crucial aspects of neural patterning in brain and spinal cord regions and along rostralcaudal and dorsalventral axes. This structure was utilized for studying neuronal lineage development, which revealed pre-patterning of axial identities of neural crest progenitors and functional roles of neuromesodermal progenitors and the caudal gene CDX2 in spinal cord and trunk neural crest development. We further developed dorsalventral patterned microfluidic forebrain-like structures with spatially segregated dorsal and ventral regions and layered apicobasal cellular organizations that mimic development of the human forebrain pallium and subpallium, respectively. Together, these microfluidics-based neurodevelopment models provide three-dimensional lumenal tissue architectures with in vivo-like spatiotemporal cell differentiation and organization, which will facilitate the study of human neurodevelopment and disease.
2023
-
(2023) Nature Communications. 14, 1, 3293. Abstract
Lissencephaly-1 (LIS1) is associated with neurodevelopmental diseases and is known to regulate the molecular motor cytoplasmic dynein activity. Here we show that LIS1 is essential for the viability of mouse embryonic stem cells (mESCs), and it governs the physical properties of these cells. LIS1 dosage substantially affects gene expression, and we uncovered an unexpected interaction of LIS1 with RNA and RNA-binding proteins, most prominently the Argonaute complex. We demonstrate that LIS1 overexpression partially rescued the extracellular matrix (ECM) expression and mechanosensitive genes conferring stiffness to Argonaute null mESCs. Collectively, our data transforms the current perspective on the roles of LIS1 in post-transcriptional regulation underlying development and mechanosensitive processes.
-
(2023) Medical Image Computing and Computer Assisted Intervention MICCAI 2023 - 26th International Conference, Proceedings. Madabhushi A., Greenspan H., Duncan J., Mousavi P., Taylor R., Salcudean S. & Syeda-Mahmood T.(eds.). p. 613-623 Abstract
Accurate 3D mitochondria instance segmentation in electron microscopy (EM) is a challenging problem and serves as a prerequisite to empirically analyze their distributions and morphology. Most existing approaches employ 3D convolutions to obtain representative features. However, these convolution-based approaches struggle to effectively capture long-range dependencies in the volume mitochondria data, due to their limited local receptive field. To address this, we propose a hybrid encoder-decoder framework based on a split spatio-temporal attention module that efficiently computes spatial and temporal self-attentions in parallel, which are later fused through a deformable convolution. Further, we introduce a semantic foreground-background adversarial loss during training that aids in delineating the region of mitochondria instances from the background clutter. Our extensive experiments on three benchmarks, Lucchi, MitoEM-R and MitoEM-H, reveal the benefits of the proposed contributions achieving state-of-the-art results on all three datasets. Our code and models are available at https://github.com/OmkarThawakar/STT-UNET.
-
(2023) BioEssays. 45, 9, 2300039. Abstract
Heterogeneous nuclear ribonucleoprotein U (HNRNPU) is a nuclear protein that plays a crucial role in various biological functions, such as RNA splicing and chromatin organization. HNRNPU/scaffold attachment factor A (SAF-A) activities are essential for regulating gene expression, DNA replication, genome integrity, and mitotic fidelity. These functions are critical to ensure the robustness of developmental processes, particularly those involved in shaping the human brain. As a result, HNRNPU is associated with various neurodevelopmental disorders (HNRNPU-related neurodevelopmental disorder, HNRNPU-NDD) characterized by developmental delay and intellectual disability. Our research demonstrates that the loss of HNRNPU function results in the death of both neural progenitor cells and post-mitotic neurons, with a higher sensitivity observed in the former. We reported that HNRNPU truncation leads to the dysregulation of gene expression and alternative splicing of genes that converge on several signaling pathways, some of which are likely to be involved in the pathology of HNRNPU-related NDD.
-
-
(2023) iScience. 26, 1, 105797. Abstract
Generating effective therapies for neurodevelopmental disorders has remained elusive. An emerging drug discovery approach for neurodevelopmental disorders is to characterize transcriptome-wide dysregulation in an appropriate model system and screen therapeutics based on their capacity to restore functionally relevant expression patterns. We characterized transcriptomic dysregulation in a human model of HNRNPU-related disorder to explore the potential of such a paradigm. We identified widespread dysregulation in functionally relevant pathways and then compared dysregulation in a human model to transcriptomic differences in embryonic and perinatal mice to determine whether dysregulation in an in vitro human model is partially replicated in an in vivo model of HNRNPU-related disorder. Strikingly, we find enrichment of co-dysregulation between 45-day-old human organoids and embryonic, but not perinatal, mice from distinct models of HNRNPU-related disorder. Thus, hnRNPU deficient human organoids may only be suitable to model transcriptional dysregulation in certain cell types within a specific developmental time window.
-
(2023) Neocortical Neurogenesis in Development and Evolution. Huttner W.(eds.). p. 365-396 Abstract
LIS1 (LISSENCEPHALY-1, also known as PAFAH1B1) is the first gene identified and reported to be involved in neuronal migration. Multiple studies have demonstrated that LIS1 is vital in regulating the critical retrograde molecular motor, cytoplasmic dynein. Recent studies show that LIS1 is an essential protein interacting with numerous others, including many RNA-interacting proteins. These findings position LIS1 as a critical hub protein involved in pleiotropic traits. This chapter will discuss how LIS1 can participate in diverse processes, from basic RNA biology to biomechanics regulation, and how its malfunction may contribute to the pathophysiology of various CNS diseases.
2022
-
(2022) Nature Communications. 13, 1, 4209. Abstract
HNRNPU encodes the heterogeneous nuclear ribonucleoprotein U, which participates in RNA splicing and chromatin organization. Microdeletions in the 1q44 locus encompassing HNRNPU and other genes and point mutations in HNRNPU cause brain disorders, including early-onset seizures and severe intellectual disability. We aimed to understand HNRNPUs roles in the developing brain. Our work revealed that HNRNPU loss of function leads to rapid cell death of both postmitotic neurons and neural progenitors, with an apparent higher sensitivity of the latter. Further, expression and alternative splicing of multiple genes involved in cell survival, cell motility, and synapse formation are affected following Hnrnpus conditional truncation. Finally, we identified pharmaceutical and genetic agents that can partially reverse the loss of cortical structures in Hnrnpu mutated embryonic brains, ameliorate radial neuronal migration defects and rescue cultured neural progenitors cell death.
-
(2022) Nature Communications. 13, 1, 633. Abstract
The choroid plexus secretes cerebrospinal fluid and is critical for the development and function of the brain. In the telencephalon, the choroid plexus epithelium arises from the Wnt- expressing cortical hem. Canonical Wnt signaling pathway molecules such as nuclear β-CATENIN are expressed in the mouse and human embryonic choroid plexus epithelium indicating that this pathway is active. Point mutations in human β-CATENIN are known to result in the constitutive activation of canonical Wnt signaling. In a mouse model that recapitulates this perturbation, we report a loss of choroid plexus epithelial identity and an apparent transformation of this tissue to a neuronal identity. Aspects of this phenomenon are recapitulated in human embryonic stem cell derived organoids. The choroid plexus is also disrupted when β-Catenin is conditionally inactivated. Together, our results indicate that canonical Wnt signaling is required in a precise and regulated manner for normal choroid plexus development in the mammalian brain.
-
(2022) Nature. 609, 7929, p. 907-910 Abstract
Self-organizing three-dimensional cellular models derived from human pluripotent stem cells or primary tissue have great potential to provide insights into how the human nervous system develops, what makes it unique and how disorders of the nervous system arise, progress and could be treated. Here, to facilitate progress and improve communication with the scientific community and the public, we clarify and provide a basic framework for the nomenclature of human multicellular models of nervous system development and disease, including organoids, assembloids and transplants.
-
(2022) Cells. 11, 10, 1642. Abstract
The cortex is a highly organized structure that develops from the caudal regions of the segmented neural tube. Its spatial organization sets the stage for future functional arealization. Here, we suggest using a developmental perspective to describe and understand the etiology of common cortical malformations and their manifestation in the human brain.
-
(2022) Frontiers in Cell and Developmental Biology. 10, 769853. Abstract
LIS1 (PAFAH1B1) plays a major role in the developing cerebral cortex, and haploinsufficient mutations cause human lissencephaly type 1. We have studied morphological and functional properties of the cerebral cortex of mutant mice harboring a deletion in the first exon of the mouse Lis1 (Pafah1b1) gene, which encodes for the LisH domain. The Lis1/sLis1 animals had an overall unaltered cortical structure but showed an abnormal distribution of cortical GABAergic interneurons (those expressing calbindin, calretinin, or parvalbumin), which mainly accumulated in the deep neocortical layers. Interestingly, the study of the oscillatory activity revealed an apparent inability of the cortical circuits to produce correct activity patterns. Moreover, the fast spiking (FS) inhibitory GABAergic interneurons exhibited several abnormalities regarding the size of the action potentials, the threshold for spike firing, the time course of the action potential after-hyperpolarization (AHP), the firing frequency, and the frequency and peak amplitude of spontaneous excitatory postsynaptic currents (sEPSCs). These morphological and functional alterations in the cortical inhibitory system characterize the Lis1/sLis1 mouse as a model of mild lissencephaly, showing a phenotype less drastic than the typical phenotype attributed to classical lissencephaly. Therefore, the results described in the present manuscript corroborate the idea that mutations in some regions of the Lis1 gene can produce phenotypes more similar to those typically described in schizophrenic and autistic patients and animal models.
-
(2022) ALTEX, alternatives to animal experimentation. 39, 4, p. 694-709 Abstract
Parkinson's disease (PD) is a complex neurodegenerative condition with a multifactorial origin. To date, approaches to drug discovery for PD have resulted in symptomatic therapies for the motor manifestations and signs associated with neurodegeneration but have failed to identify preventive or curative therapies. This failure mainly originates from the persistence of major gaps in our understanding of the specific molecular basis of PD initiation and progression. New approach methodologies (NAMs) hold the potential to advance PD research while facilitating a move away from animal-based research. We report a workshop involving NAM experts in the field of PD and neurodegenerative diseases, who discussed and identified a scientific strategy for successful, human-specific PD research. We outline some of the most important human-specific NAMs, along with their main potentials and limitations, and suggest possible ways to overcome the latter. Key recommendations to advance PD research include integrating NAMs while accounting for multiple levels of complexity, from molecular to population level; learning from recent advances in Alzheimer's disease research; increasing the sharing of data; promoting innovative pilot studies on disease pathogenesis; and accessing philanthropic funding to enable studies using novel approaches. Collaborative efforts between different stakeholders including researchers, clinicians and funding agencies are urgently needed to create a scientific roadmap and support a paradigm change towards effective, human-specific research for neurodegenerative diseases without animals, as is already happening in the field of toxicology.
2021
-
(2021) Frontiers in Immunology. 12, 694810. Abstract
First Paragraph:The complement system is an evolutionarily ancient arm of the innate immune system. It is composed of over 40 proteins, receptors and regulators that interact in a cascade manner to protect the host against pathogens (1). The soluble circulating complement proteins are mainly produced by hepatocytes. However, it is now well established that complement factors are expressed throughout the body, including in the central nervous system (CNS). In recent years, complement factors have been shown to control major aspects of CNS development, health, injury and disease (24). This Research Topic gathers the latest contributions to how complement factors interact with the nervous system, providing new mechanistic insight into neurodevelopment, cognitive function, myelination, and CNS infection. Review articles highlight roles for complement in development and degeneration of the visual system, and also the status of translation and clinical trials for complement-targeted therapeutics.
-
(2021) Current Opinion in Neurobiology. 66, p. 30-36 Abstract
During the last few decades we have witnessed an impressive gain in the knowledge regarding the basic mechanisms underlying human neuronal migration disorders by the usage of mouse models. Nevertheless, despite the remarkable conservation both in the genetic encoded information and the developmental processes, there are still numerous important differences between human and mouse. This may explain the vast excitement following the realization that technological breakthroughs enabled generating tissue-like human-based organoids for modeling human neuronal migration diseases. This review will provide a short introduction on human and mouse neuronal migration processes, and highlight human brain organoid models of neuronal migration diseases.
-
(2021) Recent Advances in iPSC Technology, Volume 5. Vol. 5. p. 157-177 Abstract
Recent advances in stem cell, genome editing, and iPSC technologies have made it possible to study human brain evolution using this iPSC-derived brain organoids as a model system.In this chapter, we will explore the evolution of the human brain and the central nervous system and discuss what we can learn from genomic comparisons studies. We will also examine the model organisms used to study gene expression and cell biology and delve into the new world of iPSC-derived brain organoids and what this new model system can tell us about human brain evolution.
2020
-
(2020) Bioengineering. 7, 4, p. 1-17 164. Abstract
Recent advances in stem-cell technologies include the differentiation of human embryonic stem cells (hESCs) into organ-like structures (organoids). These organoids exhibit remarkable self-organization that resembles key aspects of in vivo organ development. However, organoids have an unpredictable anatomy, and poorly reflect the topography of the dorsoventral, mediolateral, and anteroposterior axes. In vivo the temporal and the spatial patterning of the developing tissue is orchestrated by signaling molecules called morphogens. Here, we used morphogen-soaked beads to influence the spatial identities within hESC-derived brain organoids. The morphogen-and synthetic molecules-soaked beads were interpreted as local organizers, and key transcription factor expression levels within the organoids were affected as a function of the distance from the bead. We used an on-chip imaging device that we have developed, that allows live imaging of the developing hESC-derived organoids. This platform enabled studying the effect of changes in WNT/BMP gradients on the expression of key landmark genes in the on-chip human brain organoids. Titration of CHIR99201 (WNT agonist) and BMP4 directed the expression of telencephalon and medial pallium genes; dorsal and ventral midbrain markers; and isthmus-related genes. Overall, our protocol provides an opportunity to study phenotypes of altered regional specification and defected connectivity, which are found in neurodevelopmental diseases.
-
(2020) Molecular Psychiatry. 26, 3, p. 725-735 Abstract
In recent years, it has been revealed that Parkinsons disease pathology may begin to manifest in the gastrointestinal track at a much earlier time point than in the brain. This paradigm shift has been suggested following evidence in humans that has been reproduced in animal models. Since rodent models cannot recapitulate many of the human disease features, human induced pluripotent stem cells derived from Parkinsons patients have been used to generate brain organoids, greatly contributing to our understanding of the disease pathophysiology. To understand the multifaced aspects of Parkinsons disease, it may be desirable to expand the complexity of these models, to include different brain regions, vasculature, immune cells as well as additional diverse organ-specific organoids such as gut and intestine. Furthermore, the contribution of gut microbiota to disease progression cannot be underestimated. Recent biotechnological advances propose that such combinations may be feasible. Here we discuss how this need can be met and propose that additional brain diseases can benefit from this approach.
-
(2020) Nature. 587, 7834, p. 377-386 Abstract
Here we describe the LifeTime Initiative, which aims to track, understand and target human cells during the onset and progression of complex diseases, and to analyse their response to therapy at single-cell resolution. This mission will be implemented through the development, integration and application of single-cell multi-omics and imaging, artificial intelligence and patient-derived experimental disease models during the progression from health to disease. The analysis of large molecular and clinical datasets will identify molecular mechanisms, create predictive computational models of disease progression, and reveal new drug targets and therapies. The timely detection and interception of disease embedded in an ethical and patient-centred vision will be achieved through interactions across academia, hospitals, patient associations, health data management systems and industry. The application of this strategy to key medical challenges in cancer, neurological and neuropsychiatric disorders, and infectious, chronic inflammatory and cardiovascular diseases at the single-cell level will usher in cell-based interceptive medicine in Europe over the next decade.
-
(2020) Nature Reviews Neurology. 16, 11, p. 618-635 Abstract
Malformations of cortical development (MCDs) are neurodevelopmental disorders that result from abnormal development of the cerebral cortex in utero. MCDs place a substantial burden on affected individuals, their families and societies worldwide, as these individuals can experience lifelong drug-resistant epilepsy, cerebral palsy, feeding difficulties, intellectual disability and other neurological and behavioural anomalies. The diagnostic pathway for MCDs is complex owing to wide variations in presentation and aetiology, thereby hampering timely and adequate management. In this article, the international MCD network Neuro-MIG provides consensus recommendations to aid both expert and non-expert clinicians in the diagnostic work-up of MCDs with the aim of improving patient management worldwide. We reviewed the literature on clinical presentation, aetiology and diagnostic approaches for the main MCD subtypes and collected data on current practices and recommendations from clinicians and diagnostic laboratories within Neuro-MIG. We reached consensus by 42 professionals from 20 countries, using expert discussions and a Delphi consensus process. We present a diagnostic workflow that can be applied to any individual with MCD and a comprehensive list of MCD-related genes with their associated phenotypes. The workflow is designed to maximize the diagnostic yield and increase the number of patients receiving personalized care and counselling on prognosis and recurrence risk.
-
(2020) Patterning and Cell Type Specification in the Developing CNS and PNS. p. 205-221 Abstract
Organoids are three-dimensional cell cultures derived from mammalian stem cells that recapitulate key aspects of embryonic organ development and morphogenesis. In particular, brain organoids are cell cultures designed to mimic aspects of embryonic brain development. These systems exhibit remarkable self-organization of shape and cell fate. Brain organoids allow us to study a variety of neurodevelopmental phenomena ranging from early developmental events such as neuronal differentiation and migration to later events such as synapse formation and network activity. Importantly, brain organoids can be derived from human origin and thus offer the possibility to study human brain developmental disorders such as lissencephaly and schizophrenia. In the first part of this chapter, we discuss the relevance of human brain organoid systems to embryonic development and review the key neurodevelopmental phenomena that have been observed in organoids. In the second part of the chapter, we provide an overview of the use of brain organoid as a platform to study and understand neurodevelopmental disorders.
-
(2020) Cellular Migration and Formation of Axons and Dendrites. p. 305-322 Abstract
Nucleokinesis, literally the movement of the nucleus, may or may not be coupled with cellular motility. In this chapter, we present our point of view on the moving nucleus. We depict the structure of the nucleus, discuss how the chromatin is organized within the nucleus, highlight nuclear substructure, and describe the molecules that anchor it to the cytoskeleton. We further discuss diseases that arise when these molecules malfunction. How the nucleus connects with the cytoskeleton is key in understanding the relationships between these two different domains. The structure and the role of the bridging complex, the LINC complex, are presented as well. As an example for nuclear motility, we focus on dynamic nucleokinesis during interkinetic nuclear movement. Collectively, we aim to introduce the concept that the migrating nucleus is more than just a passive cargo.
-
(2020) Frontiers in Neuroscience. 14, 23. Abstract
Current evidence indicates that certain immune molecules such as components of the complement system are directly involved in neurobiological processes related to brain development, including neurogenesis, neuronal migration, synaptic remodeling, and response to prenatal or early postnatal brain insults. Consequently, complement system dysfunction has been increasingly implicated in disorders of neurodevelopmental origin, such as schizophrenia, autism spectrum disorder (ASD) and Rett syndrome. However, the mechanistic evidence for a causal relationship between impaired complement regulation and these disorders varies depending on the disease involved. Also, it is still unclear to what extent altered complement expression plays a role in these disorders through inflammation-independent or -dependent mechanisms. Furthermore, pathogenic mutations in specific complement components have been implicated in the etiology of 3MC syndrome, a rare autosomal recessive developmental disorder. The aims of this review are to discuss the current knowledge on the roles of the complement system in sculpting brain architecture and function during normal development as well as after specific inflammatory insults, such as maternal immune activation (MIA) during pregnancy, and to evaluate the existing evidence associating aberrant complement with developmental brain disorders.
2019
-
(2019) Molecular Psychiatry. 26, 5, p. 1535-1550 Abstract
The broad impairments in cognitive and neurologic functioning found in Autism Spectrum Disorder (ASD) patients are thought to originate during early prenatal developmental stages. Indeed, postmortem and imaging studies in ASD patients detected white-matter abnormalities, as well as prefrontal and temporal cortex deficits, evident from early childhood. Here, we used Maternal Immune Activation (MIA), a mouse model for ASD, in which the offsprings exhibit Autistic-like behaviors as well as cortical abnormalities. However, the dynamics that influence the number and the identity of newly born cortical neurons following maternal inflammation remains unknown. Our study shows early changes in the duration of the S-phase of PAX6+ progenitors, leading to an increased proportion of neurogenic divisions and a reciprocal decrease in the proliferative divisions. In two different time points of maternal inflammation, MIA resulted in an overproduction of CTIP2+ cortical neurons, which remained overrepresented at the end of gestation and in postnatal mice. Interestingly, MIA-resistant IL6-KO mice did not exhibit these changes. Lastly, we propose that elevated levels of the transcription factor PAX6 following MIA supports the overproduction of CTIP2+ neurons. Taken together, our data reveals a possible link between maternal immune activation and the excess of cortical neurons found in the cortex of ASD patients.
-
(2019) FRONTIERS IN CELLULAR NEUROSCIENCE. 13, 434. Abstract
In the middle of March 2019, a group of scientists and clinicians (as well as those who wear both hats) gathered in the green campus of the Weizmann Institute of Science to share recent scientific findings, to establish collaborations, and to discuss future directions for better diagnosis, etiology modeling and treatment of brain malformations. One hundred fifty scientists from twenty-two countries took part in this meeting. Thirty-eight talks were presented and as many as twenty-five posters were displayed. This review is aimed at presenting some of the highlights that the audience was exposed to during the three-day meeting.
-
(2019) FRONTIERS IN CELLULAR NEUROSCIENCE. 13, 370. Abstract
LIS1 is the main causative gene for lissencephaly, while MeCP2 is the main causative gene for Rett syndrome, both of which are neurodevelopmental diseases. Here we report nuclear functions for LIS1 and identify previously unrecognized physical and genetic interactions between the products of these two genes in the cell nucleus, that has implications on MeCP2 organization, neuronal gene expression and mouse behavior. Reduced LIS1 levels affect the association of MeCP2 with chromatin. Transcriptome analysis of primary cortical neurons derived from wild type, Lis1±, MeCP2−/y, or double mutants mice revealed a large overlap in the differentially expressed (DE) genes between the various mutants. Overall, our findings provide insights on molecular mechanisms involved in the neurodevelopmental disorders lissencephaly and Rett syndrome caused by dysfunction of LIS1 and MeCP2, respectively.
-
(2019) FRONTIERS IN CELLULAR NEUROSCIENCE. 13, 92. Abstract
Palmitoyl-protein thioesterase 1 (PPT1) is a depalmitoylation enzyme that is mutated in cases of neuronal ceroid lipofuscinosis (NCL). The hallmarks of the disease include progressive neurodegeneration and blindness, as well as seizures. In the current study, we identified 62 high-confident PPT1-binding proteins. These proteins included a self-interaction of PPT1, two V-type ATPases, calcium voltage-gated channels, cytoskeletal proteins and others. Pathway analysis suggested their involvement in seizures and neuronal morphology. We then proceeded to demonstrate that hippocampal neurons from Ppt1−/− mice exhibit structural deficits, and further investigated electrophysiology parameters in the hippocampi of mutant mice, both in brain slices and dissociated postnatal primary cultures. Our studies reveal new mechanistic features involved in the pathophysiology of this devastating neurodegenerative disease.
-
(2019) PLoS ONE. 14, 2, 0212970. Abstract
Human endogenous retroviruses are remnants of ancient germline infections that make up approximately 8% of the modern human genome. The HERV-K (HML-2) family is one of the most recent entrants into the human germline, these viruses appear to be transcriptionally active, and HERV-K viral like particles (VLPs) are found in cell lines from a number of human malignancies. HERV-K VLPs were first found to be produced in teratocarcinoma cell lines, and since then teratocarcinoma has been thought of as the classical model for HERVKs, with the NCCIT teratocarcinoma cell line particularly known to produce VLPs. Treatment for teratocarcinoma has progressed since its discovery, with improved prognosis for patients. Since the introduction of platinum based therapy, first year survival has greatly improved even with disseminated disease; however, it is estimated that 20% to 30% of patients present with metastatic germ cell tumor relapse following initial treatments. Also, the toxicity associated with the use of chemotherapeutic agents used to treat germ cell tumors is still a major concern. In this study, we show that the depletion of the HERV-K accessory protein Np9 increases the sensitivity of NCCIT teratocarcinoma cells to bleomycin and cisplatin. While decreasing the expression of Np9 had only a modest effect on the baseline viability of the cells, the reduced expression of Np9 increased the sensitivity of the teratocarcinoma cells to environmental (serum starvation) and chemical (chemotherapeutic) stresses. Np9 is also essential to the migration of NCCIT teratocarcinoma cells: in a wound closure assay, reduced expression of Np9 resulted in cells migrating into the wound at a slower rate, whereas reintroduction of Np9 resulted in NCCIT cells migrating back into the wound in a manner similar to the control. These findings support the implication that the HERV-K accessory protein Np9 has oncogenic potential.
-
(2019) Bioengineering. 6, 1, 9. Abstract
Brain organoids have recently emerged as a three-dimensional tissue culture platform to study the principles of neurodevelopment and morphogenesis. Importantly, brain organoids can be derived from human stem cells, and thus offer a model system for early human brain development and human specific disorders. However, there are still major differences between the in vitro systems and in vivo development. This is in part due to the challenge of engineering a suitable culture platform that will support proper development. In this review, we discuss the similarities and differences of human brain organoid systems in comparison to embryonic development. We then describe how organoids are used to model neurodevelopmental diseases. Finally, we describe challenges in organoid systems and how to approach these challenges using complementary bioengineering techniques.
2018
-
(2018) Current Protocols in Cell Biology. 81, 1, e62. Abstract
Brain organoids are an emerging technique for studying human neurodevelopment in vitro, with biomedical implications. However, three-dimensional tissue culture poses several challenges, including lack of nutrient exchange at the organoid core and limited imaging accessibility of whole organoids. Here we present a method for culturing organoids in a micro-fabricated device that enables in situ real-time imaging over weeks with efficient nutrient exchange by diffusion. Our on-chip approach offers a means for studying the dynamics of organoid development, cell differentiation, cell cycle, and motion.
-
(2018) Opera Medica et Physiologica. 4, 2, p. 63-70 Abstract
The convoluted human cerebral cortex is one of the key features that allows for an increased neuronal density packing essential for the complex cognitive and socioemotional behaviours man possesses. Nevertheless, the underlying mechanisms involved in cortical folding remained a both intriguing and functionally important enigma. A crucial component known to be involved in the formation and maintenance of all tissues is the extracellular matrix (ECM), providing scaffolds which tie tissues and organs in place. The composition of the ECM in both developing and mature structures is constantly remodelled, degraded and secreted by numerous types of cells, and its role as a source of growth factors and signalling in morphogenesis, migration, and proliferation is increasingly appreciated. Evidence for the differential expression of ECM during gyrification pinpoints its potentially fundamental role in shaping the folds of the cerebral cortex through both mechanical and molecular configurations. This review aims at addressing key ideas, potential directions and discoveries that highlight biomechanics of the ECM during the construction of the cortex cerebral gyrification.
-
(2018) Cerebral Cortex. 28, 9, p. 3115-3128 Abstract
The evolutionarily conserved Notch pathway plays an important role in regulation of stem cell renewal and cell fate determination in numerous organs, and as such is a key pathway in normal health and disease processes. Canonical Notch signaling is usually activated by cell contact where transmembrane ligands such as Delta-like and Jagged bind to Notch receptors. Notch activation results in the translocation of the cleaved Notch intracellular domain (NICD) into the nucleus and subsequent activation of transcription. Poly-ubiquitination leading to proteosome degradation of pathway components is one mean of regulating the Notch pathway. Here, we identified that Shootin1 exhibits the surprising propensity of activating the pathway either by interacting with LNX1/2 and promoting poly-ubiquitination of Numb or by complexing with Itch and impairing poly-ubiquitination of NICD. Within the developing brain Shootin1 modulates neuroblasts cell fate by executing 2 opposing activities on ubiquitin ligases, which control Notch signaling on 2 different levels.
-
(2018) Journal of Neurochemistry. 146, 5, p. 500-525 Abstract
The cerebral cortex is a highly organized structure whose development depends on diverse progenitor cell types, namely apical radial glia, intermediate progenitors, and basal radial glia cells, which are responsible for the production of the correct neuronal output. In recent years, these progenitor cell types have been deeply studied, particularly basal radial glia and their role in cortical expansion and gyrification. We review here a broad series of factors that regulate progenitor behavior and daughter cell fate. We first describe the different neuronal progenitor types, emphasizing the differences between lissencephalic and gyrencephalic species. We then review key factors shown to influence progenitor proliferation versus differentiation, discussing their roles in progenitor dynamics, neuronal production, and potentially brain size and complexity. Although spindle orientation has been considered a critical factor for mode of division and daughter cell output, we discuss other features that are emerging as crucial for these processes such as organelle and cell cycle dynamics. Additionally, we highlight the importance of adhesion molecules and the polarity complex for correct cortical development. Finally, we briefly discuss studies assessing progenitor multipotency and its possible contribution to the production of specific neuronal populations. This review hence summarizes recent aspects of cortical progenitor cell biology, and pinpoints emerging features critical for their behavior.
-
Human brain organoids on a chip to study development and disease(2018) Opera Medica et Physiologica. 4, p. 12 Abstract
-
(2018) Frontiers in Molecular Neuroscience. 11, 150. Abstract
The complement system, which is part of the innate immune response system, has been recently shown to participate in multiple key processes in the developing brain. Here we aimed to elucidate downstream signaling responses linking complement C3, a key molecule of the pathway, to small GTPases, known to affect the cytoskeleton. The expression pattern of the activated small GTPase Rac1 resembled that of complement C3. C3-deficient mice exhibited reduced Rac1 and elevated RhoA activity in comparison with control mice. The most pronounced reduction of Rac1 activity occurred at embryonic day 14. Rac1 has been implicated in neuronal migration as well as neuronal stem cell proliferation and differentiation. Consistent with the reduction in Rac1 activity, the expression of phospho-cofilin, decreased in migrating neurons. Reduced Rac1-GTP was also correlated with a decrease in the expression of progenitor markers (Nestin, Pax6 and Tbr2) and conversely the expression of neuronal markers (Dcx and NeuN) increased in C3 knockout (KO) cortices in comparison with wild-type (WT) cortices. More specifically, C3 deficiency resulted in a reduction in the number of the cells in S-phase and an elevation in the number of cells that precociously exited the cell cycle. Collectively, our findings suggest that C3 impacts the activity of small GTPases resulting in cell cycle defects and premature neuronal differentiation.
-
(2018) Nature Physics. 14, 5, p. 515-522 Abstract
Human brain wrinkling has been implicated in neurodevelopmental disorders and yet its origins remain unknown. Polymer gel models suggest that wrinkling emerges spontaneously due to compression forces arising during differential swelling, but these ideas have not been tested in a living system. Here, we report the appearance of surface wrinkles during the in vitro development and self-organization of human brain organoids in a microfabricated compartment that supports in situ imaging over a timescale of weeks. We observe the emergence of convolutions at a critical cell density and maximal nuclear strain, which are indicative of a mechanical instability. We identify two opposing forces contributing to differential growth: cytoskeletal contraction at the organoid core and cell-cycle-dependent nuclear expansion at the organoid perimeter. The wrinkling wavelength exhibits linear scaling with tissue thickness, consistent with balanced bending and stretching energies. Lissencephalic (smooth brain) organoids display reduced convolutions, modified scaling and a reduced elastic modulus. Although the mechanism here does not include the neuronal migration seen in vivo, it models the physics of the folding brain remarkably well. Our on-chip approach offers a means for studying the emergent properties of organoid development, with implications for the embryonic human brain.
-
(2018) Cell Stem Cell. 22, 1, p. 3-4 Abstract
In this issue of Cell Stem Cell, Jinnou et al. (2018) identify a limited time window wherein neonatal brain injuries may be treated through neuroblast migration toward the injury site on radial glial fibers. Implanting a sponge coated with an adhesive factor in the injured neonatal brain supports the migration of neuroblasts and improves functional recovery.
2017
-
(2017) FRONTIERS IN CELLULAR NEUROSCIENCE. 11, 169. Abstract
Current knowledge regarding regulation of radial neuronal migration is mainly focused on intracellular molecules. Our unbiased screen aimed at identification of non-cell autonomous mechanisms involved in this process detected differential expression of Serping1 or C1 inhibitor, which is known to inhibit the initiation of the complement cascade. The complement cascade is composed of three pathways; the classical, lectin, and the alternative pathway; the first two are inhibited by C1 inhibitor, and all three converge at the level of C3. Knockdown or knockout of Serping1 affected neuronal stem cell proliferation and impaired neuronal migration in mice. Knockdown of Serping1 by in utero electroporation resulted in a migration delay of the electroporated cells as well as their neighboring cells demonstrating a non-cell autonomous effect. Cellular polarity was also affected. Most importantly, expression of protein components mimicking cleaved C3 rescued the knockdown of Serping1, indicating complement pathway functionality. Furthermore, we propose that this activity is mediated mainly via the complement peptide C5a receptors. Whereas addition of a selective C3a receptor agonist was minimally effective, the addition of a dual C3aR/C5a receptor agonist significantly rescued Serping1 knockdown-mediated neuronal migration defects. Our findings suggest that modulating Serping1 levels in the developing brain may affect the complement pathway in a complex way. Collectively, our findings demonstrate an unorthodox activity for the complement pathway during brain development.
-
(2017) Human Molecular Genetics. 26, 9, p. 1678-1693 Abstract
Mutations in the depalmitoylation enzyme, palmitoyl protein thioesterase (PPT1), result in the early onset neurodegenerative disease known as Infantile Neuronal Ceroid Lipofuscinosis. Here, we provide proteomic evidence suggesting that PPT1 deficiency could be considered as a ciliopathy. Analysis of membrane proteins from brain enriched for acylated proteins from neonate Ppt1 knock out and control mice revealed a list of 88 proteins with differential expression levels. Amongst them, we identified Rab3IP, which regulates ciliogenesis in concert with Rab8 and Rab11. Immunostaining analysis revealed that PPT1 is localized in the cilia. Indeed, an unbiased proteomics analysis on isolated cilia revealed 660 proteins, which differed in their abundance levels between wild type and Ppt1 knock out. We demonstrate here that Rab3IP, Rab8 and Rab11 are palmitoylated, and that palmitoylation of Rab11 is required for correct intracellular localization. Cells and brain preparations from Ppt1-/- mice exhibited fewer cells with cilia and abnormally longer cilia, with both acetylated tubulin and Rab3IP wrongly distributed along the length of cilia. Most importantly, the analysis revealed a difference in the distribution and levels of the modified proteins in cilia in the retina of mutant mice versus the wildtype, which may be important in the early neurodegenerative phenotype. Overall, our results suggest a novel link between palmitoylated proteins, cilial organization and the pathophysiology of Neuronal Ceroid Lipofuscinosis.
-
(2017) Nature Communications. 8, 15096. Abstract
In recent years the notion that malfunctioning of the immune system may result in developmental brain diseases has emerged. However, the role of immune molecules in the developing brain has not been well explored. The complement pathway converges to cleave C3. Here we show that key proteins in the lectin arm of this pathway, MASP1, MASP2 and C3, are expressed in the developing cortex and that neuronal migration is impaired in knockout and knockdown mice. Molecular mimics of C3 cleavage products rescue the migration defects that have been seen following knockdown of C3 or Masp2. Pharmacological activation of the downstream receptors rescue Masp2 and C3 knockdown as well as C3 knockout. Therefore, we propose that the complement pathway is functionally important in migrating neurons of the developing cortex.
2016
-
(2016) Development Growth and Differentiation. 58, 5, p. 481-491 Abstract
The opportunity to model autism spectrum disorders (ASD) through generation of patient-derived induced pluripotent stem cells (iPSCs) is currently an emerging topic. Wide-scale research of altered brain circuits in syndromic ASD, including Rett Syndrome, Fragile X Syndrome, Angelman's Syndrome and sporadic Schizophrenia, was made possible through animal models. However, possibly due to species differences, and to the possible contribution of epigenetics in the pathophysiology of these diseases, animal models fail to recapitulate many aspects of ASD. With the advent of iPSCs technology, 3D cultures of patient-derived cells are being used to study complex neuronal phenotypes, including both syndromic and nonsyndromic ASD. Here, we review recent advances in using iPSCs to study various aspects of the ASD neuropathology, with emphasis on the efforts to create in vitro model systems for syndromic and nonsyndromic ASD. We summarize the main cellular activity phenotypes and aberrant genetic interaction networks that were found in iPSC-derived neurons of syndromic and nonsyndromic autistic patients.
-
(2016) Journal of Neurochemistry. 136, 3, p. 440-456 Abstract
Autism spectrum disorders (ASD) encompass a group of neurodevelopmental diseases that demonstrate strong heritability, however, the inheritance is not simple and many genes have been associated with these disorders. ASD is regarded as a neurodevelopmental disorder, and abnormalities at different developmental stages are part of the disease etiology. This review provides a general background on neuronal migration during brain development and discusses recent advancements in the field connecting ASD and aberrant neuronal migration. We propose that neuronal migration impairment may be an important common pathophysiology in autism spectrum disorders (ASD). This review provides a general background on neuronal migration during brain development and discusses recent advancements in the field connecting ASD and aberrant neuronal migration. We propose that neuronal migration impairment may be an important common pathophysiology in autism spectrum disorders (ASD). This review provides a general background on neuronal migration during brain development and discusses recent advancements in the field connecting ASD and aberrant neuronal migration.
-
(2016) PLoS ONE. 11, 1, e0146466. Abstract
Mutations in the depalmitoylating enzyme gene, PPT1, cause the infantile form of Neuronal Ceroid Lipofuscinosis (NCL), an early onset neurodegenerative disease. During recent years there have been different therapeutic attempts including enzyme replacement. Here we show that PPT1 is palmitoylated in vivo and is a substrate for two palmitoylating enzymes, DHHC3 and DHHC7. The palmitoylated protein is detected in both cell lysates and medium. The presence of PPT1 with palmitoylated signal peptide in the cell medium suggests that a subset of the protein is secreted by a nonconventional mechanism. Using a mutant form of PPT1, C6S, which was not palmitoylated, we further demonstrate that palmitoylation does not affect intracellular localization but rather that the unpalmitoylated form enhanced the depalmitoylation activity of the protein. The calculated Vmax of the enzyme was significantly affected by the palmitoylation, suggesting that the addition of a palmitate group is reminiscent of adding a noncompetitive inhibitor. Thus, we reveal the existence of a positive feedback loop, where palmitoylation of PPT1 results in decreased activity and subsequent elevation in the amount of palmitoylated proteins. This positive feedback loop is likely to initiate a vicious cycle, which will enhance disease progression. The understanding of this process may facilitate enzyme replacement strategies.
2015
-
(2015) Frontiers in Neuroscience. 9, MAR, 53. Abstract
The intricate formation of the cerebral cortex requires a well-coordinated series of events, which are regulated at the level of cell-autonomous and non-cell autonomous mechanisms. Whereas cell-autonomous mechanisms that regulate cortical development are well-studied, the non-cell autonomous mechanisms remain poorly understood. A non-biased screen allowed us to identify Autotaxin (ATX) as a non-cell autonomous regulator of neural stem cells. ATX (also known as ENPP2) is best known to catalyze lysophosphatidic acid (LPA) production. Our results demonstrate that ATX affects the localization and adhesion of neuronal progenitors in a cell autonomous and non-cell autonomous manner, and strikingly, this activity is independent from its catalytic activity in producing LPA.
-
(2015) Journal of Neuroscience. 35, 3, p. 936-942 Abstract
Spinal muscular atrophy with pontocerebellar hypoplasia (SMA-PCH) is an infantile SMA variant with additional manifestations, particularly severe microcephaly. We previously identified a nonsense mutation in Vaccinia-related kinase 1 (VRK1), R358X, as a cause of SMA-PCH. VRK1-R358X is a rare founder mutation in Ashkenazi Jews, and additional mutations in patients of different origins have recently been identified.VRK1is a nuclear serine/threonine protein kinaseknownto play multiple roles in cellular proliferation, cell cycle regulation, and carcinogenesis. However, VRK1 was not known to have neuronal functions before its identification as a gene mutated in SMA-PCH. Here we show that VRK1-R358X homozygosity results in lack of VRK1 protein, and demonstrate a role for VRK1 in neuronal migration and neuronal stem cell proliferation. Using shRNA in utero electroporation in mice, we show that Vrk1 knockdown significantly impairs cortical neuronal migration, and affects the cell cycle of neuronal progenitors. Expression of wild-type human VRK1 rescues both proliferation and migration phenotypes. However, kinase-dead human VRK1 rescues only the migration impairment, suggesting the role of VRK1 in neuronal migration is partly noncatalytic. Furthermore, we found that VRK1 deficiency in human and mouse leads to downregulation of amyloid-β precursor protein (APP), a known neuronal migration gene. APP overexpression rescues the phenotype caused by Vrk1 knockdown, suggesting that VRK1 affects neuronal migration through an APP-dependent mechanism.
2014
-
(2014) Stem Cells. 32, 10, p. 2657-2667 Abstract
Emergence of genomic instability is a practical issue in preparing neural stem cells (NSCs) and induced pluripotent stem cells (iPSCs). However, it is still not fully understood what the origins and mechanisms for formation are for the genomic alternations observed. Here, we studied the extent of genomic variation on the scale of individual cells originating from the same animal. We used mouse NSCs grown from embryonic cells and iPSCs generated from embryonic brain cells, B cells or fibroblasts, and performed comparative analysis with cultures of fibroblasts from the same mouse. In the first passage of these cell lines, aneuploidies were only observed for chromosomes 6, 11, 12, 19, and Y, which is overall at a rate lower than previously reported; de novo copy number variations (CNVs) were observed in 4.3% of neural iPSCs, 29% of B cell iPSCs, 10% of fibroblast iPSCs, and 1.3% of neurospheres. In contrast, propagation of these first passage cells to a later passage induced additional aneuploidies and CNVs. Breakpoint sequencing analysis suggested that the majority of the detected CNVs arose by replicative mechanisms. Interestingly, we detected identical de novo CNVs in different single cell colonies that appeared to have arisen independently from each other, which suggests a novel CNV formation mechanism in these cells. Our findings provide insights into mechanisms of CNV formation during reprogramming and suggest that replicative mechanisms for CNV formation accompany mitotic divisions.
-
(2014) Cellular and Molecular Control of Neuronal Migration. p. 97-111 Abstract
Proper brain development requires the orchestrated migration of neurons from their place of birth to their final positioning, where they will form appropriate connections with their target cells. These events require coordinated activity of multiple elements of the cytoskeleton, in which the MARK/Par-1 polarity kinase plays an important role. Here, the various roles and modes of regulation of MARK/Par-1 are reviewed. MARK/Par-1 participates in axon formation in primary hippocampal neurons. Balanced levels of MARK/Par-1 are required for proper radial migration, as well as for migration in the rostral migratory stream. Normal neuronal migration requires at least two of MARK/Par-1 substrates, DCX and tau. Overall, the positioning of MARK/Par-1 at the crosstalk of regulating cytoskeletal dynamics allows its participation in neuronal polarity decisions.
2013
-
(2013) Current Opinion in Neurobiology. 23, 6, p. 951-956 Abstract
LIS1, the first gene to be identified as involved in a neuronal migration disease, is a dosage-sensitive gene whose proper levels are required for multiple aspects of cortical development. Deletions in LIS1 result in a severe brain malformation, known as lissencephaly, whereas duplications delay brain development. LIS1 affects the proliferation of progenitors, spindle orientation and interkinetic nuclear movement in the ventricular zone, as well as nucleokinesis and migration of neurons. LIS1 regulatory interaction with the minus end directed molecular motor cytoplasmic dynein is the key for understanding its complex cellular functions. LIS1-dynein interaction decreases the average velocity of the molecular motor in vitro, shows more complex effects in vivo, and may be of importance in high-load transport especially in neurons.
-
(2013) Journal of visualized experiments : JoVE. 79, p. e50146 Abstract
In utero electroporation (IUE) is a technique which allows genetic modification of cells in the brain for investigating neuronal development. So far, the use of IUE for investigating behavior or neuropathology in the adult brain has been limited by insufficient methods for monitoring of IUE transfection success by non-invasive techniques in postnatal animals. For the present study, E16 rats were used for IUE. After intraventricular injection of the nucleic acids into the embryos, positioning of the tweezer electrodes was critical for targeting either the developing cortex or the hippocampus. Ventricular co-injection and electroporation of a luciferase gene allowed monitoring of the transfected cells postnatally after intraperitoneal luciferin injection in the anesthetized live P7 pup by in vivo bioluminescence, using an IVIS Spectrum device with 3D quantification software. Area definition by bioluminescence could clearly differentiate between cortical and hippocampal electroporations and detect a signal longitudinally over time up to 5 weeks after birth. This imaging technique allowed us to select pups with a sufficient number of transfected cells assumed necessary for triggering biological effects and, subsequently, to perform behavioral investigations at 3 month of age. As an example, this study demonstrates that IUE with the human full length DISC1 gene into the rat cortex led to amphetamine hypersensitivity. Co-transfected GFP could be detected in neurons by post mortem fluorescence microscopy in cryosections indicating gene expression present at ≥6 months after birth. We conclude that postnatal bioluminescence imaging allows evaluating the success of transient transfections with IUE in rats. Investigations on the influence of topical gene manipulations during neurodevelopment on the adult brain and its connectivity are greatly facilitated. For many scientific questions, this technique can supplement or even replace the use of transgenic rats and provide a novel technology for behavioral neuroscience.
-
(2013) Journal of Neuroscience. 33, 29, p. 11932-11948 Abstract
Shootin1 has been ascribed a role in regulating polarization of primary hippocampal neurons. To better understand the possible role of Shootin1 in the developing brain, we identified a member of the kinesin superfamily, KIF20B, as a novel Shootin1 interacting protein and a potential mediator of Shootin1 interaction with microtubules. KIF20B/Shootin1 binding was mapped to a 57 aa KIF20B sequence, which was used as a dominant-negative fragment. Direct interaction between that peptide (MBD) and Shootin1 was confirmed by surface plasmon resonance-based technology and the affinity was determined in the 10(-7) M range. The proteins are expressed in the developing brain and formed a complex in vivo based on coimmunoprecipitation experiments and coimmunostaining in primary neurons. In primary hippocampal neurons Kif20b knockdown reduced Shootin1 mobilization to the developing axon, as evidenced by immunostaining and fluorescence recovery after photobleaching analysis, suggesting that Shootin1 is a novel KIF20B cargo. shRNA targeting of Shootin1 reduced PIP3 accumulation in the growth cone, as did Kif20b shRNA. In the developing mouse brain, Kif20b knockdown or expression of the KIF20B minimal binding domain inhibited neuronal migration, and in vivo migration assays suggested that Shootin1/Kif20b acts in the same genetic pathway. Time-lapse imaging of multipolar cells in the subventricular zone revealed that downregulating levels of either Shootin1 or Kif20b hindered the transition from multipolar to bipolar cells. Collectively, our data demonstrate the importance of the Shootin1/KIF20B interaction to the dynamic process of pyramidal neuronal polarization and migration.
-
-
(2013) Cellular and Molecular Life Sciences. 70, 7, p. 1255-1268 Abstract
Microtubules are known to drive chromosome movements and to induce nuclear envelope breakdown during mitosis and meiosis. Here we show that microtubules can enforce nuclear envelope folding and alter the levels of nuclear envelope-associated heterochromatin during interphase, when the nuclear envelope is intact. Microtubule reassembly, after chemically induced depolymerization led to folding of the nuclear envelope and to a transient accumulation of condensed chromatin at the site nearest the microtubule organizing center (MTOC). This microtubule-dependent chromatin accumulation next to the MTOC is dependent on the composition of the nuclear lamina and the activity of the dynein motor protein. We suggest that forces originating from simultaneous polymerization of microtubule fibers deform the nuclear membrane and the underlying lamina. Whereas dynein motor complexes localized to the nuclear envelope that slide along the microtubules transfer forces and/or signals into the nucleus to induce chromatin reorganization and accumulation at the nuclear membrane folds. Thus, our study identified a molecular mechanism by which mechanical forces generated in the cytoplasm reshape the nuclear envelope, alter the intranuclear organization of chromatin, and affect the architecture of the interphase nucleus.
2012
-
(2012) Journal of Neuroscience. 32, 50, p. 18204-18214 Abstract
Amyloid-β peptide (Aβ) is believed to play a central role in the pathogenesis of Alzheimer's disease. In view of the side effects associated with inhibiting the secretases that produce Aβ, new molecular targets are required to provide alternative therapeutic options. We used RNA interference (RNAi) to systematically screen the Drosophila genome to identify genes that modulate Aβ production upon knockdown. RNAi of 41 genes in Drosophila cells significantly lowered Aβ without affecting general secretion or viability. After the γ-secretase complex components, the most potent effect was observed for platelet activating factor acetylhydrolase α (Paf-AHα), and, in mammalian cells, the effect was replicated for its ortholog PAFAH1B2. Knockdown of PAFAH1B2 strongly reduced Aβ secretion from human cells, and this effect was confirmed in primary cells derived from PAFAH1B2 knock-out mice. Reduced Aβ production was not attributable to altered β-amyloid precursor protein (APP) ectodomain shedding but was a result of an enhanced degradation of APP C-terminal fragments (CTFs) in the absence of PAFAH1B2 but not its close homolog PAFAH1B3. Enhanced degradation of APP CTFs was selective because no such effects were obtained for Notch or E-/N-cadherin. Thus, we have identified an important protein that can selectively modify Aβ generation via a novel mechanism, namely enhanced degradation of its immediate precursor. In view of the absence of a neurological phenotype in PAFAH1B2 knock-out mice, targeted downregulation of PAFAH1B2 may be a promising new strategy for lowering Aβ.
-
-
(2012) Human Molecular Genetics. 21, 8, p. 1681-1692 ddr603. Abstract
Microdeletions encompassing the MAPT (Tau) locus resulting in intellectual disability raised the hypothesis that Tau may regulate early functions in the developing brain. Our results indicate that neuronal migration was inhibited in mouse brains following Tau reduction. In addition, the leading edge of radially migrating neurons was aberrant in spite of normal morphology of radial glia. Furthermore, intracellular mitochondrial transport and morphology were affected. In early postnatal brains, a portion of Tau knocked down neurons reached the cortical plate. Nevertheless, they exhibited far less developed dendrites and a striking reduction in connectivity evident by the size of boutons. Our novel results strongly implicate MAPT as a dosage-sensitive gene in this locus involved in intellectual disability. Furthermore, our results are likely to impact our understanding of other diseases involving Tau.
-
(2012) Journal of Molecular Neuroscience. 46, 3, p. 516-526 Abstract
The nuclei of neuroepithelial cells move along the apicobasal axis in synchronization with their cell cycle status. This motility is known as interkinetic nuclear movement. We discuss here the importance of cytoskeleton organization, the centrosome, molecular motors, cell polarity proteins, and their regulators in controlling and maintaining this typical behavior. Furthermore, due to the tight linkage between cell proliferation, cell cycle, and nuclear motility, we speculate that interkinetic nuclear movement is likely to be affected in the pathophysiology of microcephaly, where the brain size is markedly reduced.
-
(2012) Biology Open. 1, 3, p. 220-231 Abstract
Bidirectional transport is a key issue in cellular biology. It requires coordination between microtubule-associated molecular motors that work in opposing directions. The major retrograde and anterograde motors involved in bidirectional transport are cytoplasmic dynein and conventional kinesin, respectively. It is clear that failures in molecular motor activity bear severe consequences, especially in the nervous system. Neuronal migration may be impaired during brain development, and impaired molecular motor activity in the adult is one of the hallmarks of neurodegenerative diseases leading to neuronal cell death. The mechanisms that regulate or coordinate kinesin and dynein activity to generate bidirectional transport of the same cargo are of utmost importance. We examined how Ndel1, a cytoplasmic dynein binding protein, may regulate nonvesicular bidirectional transport. Soluble Ndel1 protein, Ndel1-derived peptides or control proteins were mixed with fluorescent beads, injected into the squid giant axon, and the bead movements were recorded using time-lapse microscopy. Automated tracking allowed for extraction and unbiased analysis of a large data set. Beads moved in both directions with a clear bias to the anterograde direction. Velocities were distributed over a broad range and were typically slower than those associated with fast vesicle transport. Ironically, the main effect of Ndel1 and its derived peptides was an enhancement of anterograde motion. We propose that they may function primarily by inhibition of dynein-dependent resistance, which suggests that both dynein and kinesin motors may remain engaged with microtubules during bidirectional transport.
-
(2012) Molecular and Cellular Neuroscience. 49, 2, p. 97-103 Abstract
In rodents and most other mammals studied, neuronal precursors generated in the subventricular zone (SVZ) migrate to the adult olfactory bulb (OB) to differentiate into interneurons called granule and periglomerular cells. How the newborn cells navigate in the postnatal forebrain to reach precisely their target area is largely unknown. However, it is often thought that postnatal neurogenesis recapitulates the neuronal development occurring during embryogenesis. During brain development, intracellular kinases are key elements for controlling cell polarization as well as the coupling between polarization and cellular movement. We show here that the polarity kinase MARK2 maintains its expression in the postnatal SVZ-OB system. We therefore investigated the potential role of this kinase in adjusting postnatal neuroblast migration. We employed mouse brain slices maintained in culture, in combination with lentiviral vector injections designed to label neuronal precursors with GFP and to diminish the expression of MARK2. Time-lapse video microscopy was used to monitor neuroblast migration in the postnatal forebrain from SVZ precursors to cells populating the OB. We found that reduced MARK2 expression resulted in altered migratory patterns and stalled neuroblasts in the rostral migratory stream (RMS). In agreement with the observed migratory defects, we report a diminution of the proportion of cells reaching the OB layers. Our study reveals the involvement of MARK2 in the maintenance of the migratory direction in postnatally-generated neuroblasts and consequently on the control of the number of newly-generated neurons reaching and integrating the appropriate target circuits.
2011
-
(2011) Journal of Cell Science. 124, 23, p. 3989-4000 Abstract
Completion of mitosis requires microtubule-dependent transport of membranes to the midbody. Here, we identified a role in cytokinesis for doublecortin domain-containing protein 5 (DCDC5), a member of the doublecortin protein superfamily. DCDC5 is a microtubuleassociated protein expressed in both specific and dynamic fashions during mitosis. We show that DCDC5 interacts with cytoplasmic dynein and Rab8 (also known as Ras-related protein Rab-8A), as well as with the Rab8 nucleotide exchange factor Rabin8 (also known as Rab-3A-interacting protein). Following DCDC5 knockdown, the durations of the metaphase to anaphase transition and cytokinesis, and the proportion of multinucleated cells increases, whereas cell viability decreases. Furthermore, knockdown of DCDC5 or addition of a dynein inhibitor impairs the entry of Golgi-complex-derived Rab8-positive vesicles to the midbody. These findings suggest that DCDC5 plays an important role in mediating dynein-dependent transport of Rab8-positive vesicles and in coordinating late cytokinesis.
-
(2011) Journal of Neuroscience. 31, 46, p. 16872-16883 Abstract
The c-Jun NH 2-terminal protein kinase (JNK), which belongs to the mitogen-activated protein kinase family, plays important roles in a broad range of physiological processes. JNK is controlled by two upstream regulators, mitogen-activated protein kinase kinase (MKK) 7 and MKK4. To elucidate the physiological functions of MKK7,we used Nestin-Cre to generate a novel mouse model in which the mkk7 gene was specifically deleted in the nervous system (Mkk7 flox/flox Nestin-Cre mice). These mice were indistinguishable from their control littermates in gross appearance during embryogenesis but died immediately after birth without breathing. Histological examination showed that the mutants had severe defects in brain development, including enlarged ventricles, reduced striatum, and minimal axon tracts. Electron microscopy revealed abnormal accumulations of filamentous structures and autophagic vacuoles in Mkk7 flox/flox Nestin-Cre brain. Further analysis showed that MKK7 deletion decreased numbers of TAG-1-expressing axons and delayed neuronal migration in the cerebrum. Neuronal differentiation was not altered. In utero electroporation studies showed that contralateral projection of axons by layer 2/3 neurons was impaired in the absence of MKK7. Moreover, MKK7 regulated axon elongation in a cell-autonomous manner in vivo, a finding confirmed in vitro. Finally, phosphorylation levels of JNK substrates, including c-Jun, neurofilament heavy chain, microtubule-associated protein 1B, and doublecortin, were reduced in Mkk7 flox/flox Nestin-Cre brain. Our findings demonstrate that the phenotype of Mkk7 flox/flox Nestin-Cre mice differs substantially from that of Mkk4 flox/flox Nestin-Cre mice, and establish that MKK7-mediated regulation of JNK is uniquely critical for both axon elongation and radial migration in the developing brain.
2010
-
(2010) Journal of Neuroscience. 30, 50, p. 16766-16776 Abstract
Axon branching plays a critical role in establishing the accurate patterning of neuronal circuits in the brain. However, the mechanisms that control axon branching remain poorly understood. Here we report that knockdown of the brain-enriched signaling protein JNK-interacting protein 3 (JIP3) triggers exuberant axon branching and self-contact in primary granule neurons of the rat cerebellar cortex. JIP3 knockdown in cerebellar slices and in postnatal rat pups in vivo leads to the formation of ectopic branches in granule neuron parallel fiber axons in the cerebellar cortex. We also find that JIP3 restriction of axon branching is mediated by the protein kinase glycogen synthase kinase 3β (GSK3β). JIP3 knockdown induces the downregulation of GSK3β in neurons, and GSK3β knockdown phenocopies the effect of JIP3 knockdown on axon branching and self-contact. Finally, we establish doublecortin (DCX) as a novel substrate of GSK3β in the control of axon branching and self-contact. GSK3β phosphorylates DCX at the distinct site of Ser327 and thereby contributes to DCX function in the restriction of axon branching. Together, our data define a JIP3-regulated GSK3β/DCX signaling pathway that restricts axon branching in the mammalian brain. These findings may have important implications for our understanding of neuronal circuitry during development, as well as the pathogenesis of neurodevelopmental disorders of cognition.
-
(2010) FRONTIERS IN CELLULAR NEUROSCIENCE. 4, MAY, 19. Abstract
Platelet-activating factor acetylhydrolase 1B (PAF-AH) inactivates the potent phospholipid platelet-activating factor (PAF) and is composed of two catalytic subunits (α1 and α2) and a dimeric regulatory subunit, LIS1. The function of the catalytic subunits in brain development remains unknown. Here we examined their effects on proliferation in the ganglionic eminences and tangential migration. In α1 and α2 catalytic subunits knockout mice we noticed an increase in the size of the ganglionic eminences resulting from increased proliferation of GABAergic neurons. Our results indicate that the catalytic subunits act as negative regulators of the Wnt signaling pathway. Overexpression of each of the PAF-AH catalytic subunits reduced the amount of nuclear beta-catenin and provoked a shift of this protein from the nucleus to the cytoplasm. In the double mutant mice, Wnt signaling increased in the ganglionic eminences and in the dorsal part of the cerebral cortex. In situ hybridization revealed increased and expanded expression of a downstream target of the Wnt pathway (Cyclin D1), and of upstream Wnt components (Tcf4, Tcf3 and Wnt7B). Furthermore, the interneurons in the cerebral cortex were more numerous and in a more advanced position. Transplantation assays revealed a non-cell autonomous component to this phenotype, which may be explained in part by increased and expanded expression of Sdf1 and Netrin-1. Our findings strongly suggest that PAF-AH catalytic subunits modulate the Wnt pathway in restricted areas of the developing cerebral cortex. We hypothesize that modulation of the Wnt pathway is the evolutionary conserved activity of the PAF-AH catalytic subunits.
-
(2010) EMBO Journal. 29, 1, p. 107-119 Abstract
Regulated activity of the retrograde molecular motor, cytoplasmic dynein, is crucial for multiple biological activities, and failure to regulate this activity can result in neuronal migration retardation or neuronal degeneration. The activity of dynein is controlled by the LIS1-Ndel1-Nde1 protein complex that participates in intracellular transport, mitosis, and neuronal migration. These biological processes are subject to tight multilevel modes of regulation. Palmitoylation is a reversible posttranslational lipid modification, which can dynamically regulate protein trafficking. We found that both Ndel1 and Nde1 undergo palmitoylation in vivo and in transfected cells by specific palmitoylation enzymes. Unpalmitoylated Ndel1 interacts better with dynein, whereas the interaction between Nde1 and cytoplasmic dynein is unaffected by palmitoylation. Furthermore, palmitoylated Ndel1 reduced cytoplasmic dynein activity as judged by Golgi distribution, VSVG and short microtubule trafficking, transport of endogenous Ndel1 and LIS1 from neurite tips to the cell body, retrograde trafficking of dynein puncta, and neuronal migration. Our findings indicate, to the best of our knowledge, for the first time that Ndel1 palmitoylation is a new mean for fine-tuning the activity of the retrograde motor cytoplasmic dynein.
2009
-
(2009) Molecular Neurobiology. 40, 1, p. 1-14 Abstract
The formation of the cerebral cortex requires migration of billions of cells from their birth position to their final destination. A motile cell must have internal polarity in order to move in a specified direction. Locomotory polarity requires the coordinated polymerization of cytoskeletal elements such as microtubules and actin combined with regulated activities of the associated molecular motors. This review is focused on migrating neurons in the developing cerebral cortex, which need to attain internal polarity in order to reach their proper target. The position and dynamics of the centrosome plays an important function in this directed motility. We highlight recent interesting findings connecting polarity proteins with neuronal migration events regulated by the microtubule-associated molecular motor, cytoplasmic dynein.
-
(2009) Cerebral Cortex. 19, SUPPL. 1, p. i42-i48 Abstract
The human genome contains only about double the number of genes in comparison to the fruit fly. This necessitates efficient recurrent usage of the same molecular components to participate in different processes. When the same proteins are used for different signaling pathways, it may be conceivable that if they go awry the phenotypic consequences may vary to a great extent. The involvement of amyloid β precursor protein, Presenilin-1, and Tau in the pathogenesis of Alzheimer's disease is well established. Here we are highlighting a second facet of their function, their participation in developmental and adult neuronal migration. We propose that the prevalent and early Anosmia found in Alzheimer's patients may be due in part to malfunctioning of the above-mentioned proteins.
-
(2009) Nature Genetics. 41, 2, p. 168-177 Abstract
Deletions of the PAFAH1B1 gene (encoding LIS1) in 17p13.3 result in isolated lissencephaly sequence, and extended deletions including the YWHAE gene (encoding 14-3-3ε) cause Miller-Dieker syndrome. We identified seven unrelated individuals with submicroscopic duplication in 17p13.3 involving the PAFAH1B1 and/or YWHAE genes, and using a 'reverse genomics' approach, characterized the clinical consequences of these duplications. Increased PAFAH1B1 dosage causes mild brain structural abnormalities, moderate to severe developmental delay and failure to thrive. Duplication of YWHAE and surrounding genes increases the risk for macrosomia, mild developmental delay and pervasive developmental disorder, and results in shared facial dysmorphologies. Transgenic mice conditionally overexpressing LIS1 in the developing brain showed a decrease in brain size, an increase in apoptotic cells and a distorted cellular organization in the ventricular zone, including reduced cellular polarity but preserved cortical cell layer identity. Collectively, our results show that an increase in LIS1 expression in the developing brain results in brain abnormalities in mice and humans.
2008
-
(2008) Cellular & Molecular Biology Letters. 13, 4, p. 614-620 Abstract
Gene trapping is used to introduce genome-wide insertional mutations in embryonic stem cells. Determining the integration site is based on high-throughput PCR, which has inevitable possibilities for mistakes, thus necessitating clone verification prior to the generation of mutant mice. Here, we propose a rapid method to validate gene identity based on the fact that many high throughput gene-trapping integrations result in fusion proteins encompassing the N-terminal portion of the gene of interest and LacZ being expressed in embryonic stem cells. Our method utilizes an immunoprecipitation assay using a specific N-terminal-directed antibody to the protein product of the gene of interest followed by a color LacZ assay of the immunoprecipitate, strongly supporting the formation of a fusion protein when the color develops.
-
(2008) Journal of Neuroscience. 28, 48, p. 13008-13013 Abstract
Abnormal neuronal migration is manifested in brain malformations such as lissencephaly. The impairment in coordinated cell motility likely reflects a faulty mechanism of cell polarization or coupling between polarization and movement. Here we report on the relationship between the polarity kinase MARK2/Par-1 and its substrate, the well-known lissencephaly-associated gene doublecortin (DCX), during cortical radial migration. We have previously shown using in utero electroporation that reduced MARK2 levels resulted in multipolar neurons stalled at the intermediate zone border, similar to the phenotype observed in the case of DCX silencing. However, whereas reduced MARK2 stabilized microtubules, we show here that knock-down of DCX increased microtubule dynamics. This led to the hypothesis that simultaneous reduction may alleviate the phenotype. Coreduction ofMARK2and DCX resulted in a partial restoration of the normal neuronal migration phenotype in vivo. The kinetic behavior of the centrosomes reflected the different molecular mechanisms activated when either protein was reduced. In the case of reducing MARK2 processive motility of the centrosome was hindered, whereas when DCX was reduced, centrosomes moved quickly but bidirectionally. Our results stress the necessity for successful coupling between the polarity pathway and cytoplasmic dynein-dependent activities for proper neuronal migration.
-
(2008) Journal of Neuroscience. 28, 22, p. 5710-5720 Abstract
Radial neuronal migration is key in structuring the layered cortex. Here we studied the role of MARK2/Par-1 in this process. The dual name stands for the MAP/microtubule affinity-regulating kinase 2 (MARK2) and the known polarity kinase 1 (Par-1). Reduced MARK2 levels using in utero electroporation resulted in multipolar neurons stalled at the intermediate zone border. Reintroduction of the wild-type kinase postmitotically improved neuronal migration. Our results indicated that reduction in MARK2 affected centrosomal dynamics in migrating neurons of the cerebral cortex. Increased MARK2 has been shown to destabilize microtubules, and here we show for the first time that reduced MARK2 stabilized microtubules in primary cultured neurons. Kinase-independent activity permitted multipolar-to-bipolar transition but did not restore proper migration. Increased MARK2 levels resulted in a different phenotype, which is loss of neuronal polarity. MARK2 kinase activity reduction hindered migration in the developing brain, which was rescued by increasing kinase activity. Our results stress the necessity of maintaining dynamic microtubules for proper neuronal migration. Furthermore, the exact requirements for MARK2 and its kinase activity vary during the course of neuronal migration. Collectively, our results stress the requirements for the different roles of MARK2 during neuronal migration.
2007
-
(2007) Developmental Neuroscience. 30, 1-3, p. 187-199 Abstract
Doublecortin (DCX) is a microtubule-associated protein necessary for neuronal migration. In spite of its ubiquitous distribution in dendrites, its possible role in dendrite development has not yet been documented. The present study examined the effects of different expression levels of DCX on the arborization of dendrites in cultured hippocampal neurons. Reduced expression of DCX following RNAi transfection resulted in reduced branch points, total length and complexity of the dendrites. Overexpression of DCX resulted in an increase in branch points and complexity of the dendrites. In contrast to control green fluorescent protein cells, DCX-overexpressing cells maintained highly branched and complex dendritic trees when subjected to reduced neuronal activity by blockade of immature GABAA receptors. These results suggest that DCX supports developing dendrites, in addition to its role in neuronal migration.
-
(2007) Traffic. 8, 11, p. 1521-1529 Abstract
Directed cell migration is a property central to multiple basic biological processes. Here, we show that directed cell migration is associated with global changes in the chromatin fiber. Polarized posttranslational changes in histone H1 along with a transient decrease in H1 mobility were detected in cells facing the scratch in a wound healing assay. In parallel to the changes in H1, the levels of the heterochromatin marker histone H3 lysine 9 tri-methylation were elevated. Interestingly, reduction of the chromatin-binding affinity of H1 altered the cell migration rates. Moreover, migration-associated changes in histone H1 were observed during nuclear motility in the simple multicellular organism Neurospora crassa . Our studies suggest that dynamic reorganization of the chromatin fiber is an early event in the cellular response to migration cues.
-
(2007) Molecular and Cellular Neuroscience. 35, 2, p. 220-229 Abstract
Mutations in the mouse Lis1 gene produce severe alterations in the developing cortex. We have examined some electrophysiological responses of cortical pyramidal neurons during the early postnatal development of Lis/sLis1 mutant mice. In P7 and P30 Lis1/sLis1 neurons we detected a lower frequency and slower decay phase of mIPSCs, and at P30 the mIPSCs amplitude and the action potential duration were reduced. Zolpidem (an agonist of GABAA receptors containing the α1 subunit) neither modified the amplitude nor the decay time of mIPSCs at P7 in Lis1/sLis1 neurons, whereas it increased the decay time at P30. The levels of GABAA receptor α1 subunit mRNA were reduced in the Lis1/sLis1 brain at P7 and P30, whereas reduced levels of the corresponding protein were only found at P7. These results demonstrate the presence of functional alterations in the postnatal Lis1/sLis1 cortex and point to abnormalities in GABAA receptor subunit switching processes during postnatal development.
2006
-
(2006) NeuroMolecular Medicine. 8, 4, p. 547-565 Abstract
Lissencephaly 1 (LIS1) was the first gene implicated in the pathogenesis of type-1 lissencephaly. More than a decade of research by multiple laboratories has revealed that LIS1 is a key node protein, which participates in several pathways, including association with the molecular motor cytoplasmic dynein, the reelin signaling pathway, and the platelet-activating factor pathway. Mutations in LIS1-interacting proteins, either in human, or in mouse models has suggested that LIS1 might play a role in the pathogenesis of numerous diseases such as male sterility, schizophrenia, neuronal degeneration, and viral infections.
-
(2006) BMC Genomics. 7, 188. Abstract
Background: Doublecortin (DCX) domains serve as protein-interaction platforms. Mutations in members of this protein superfamily are linked to several genetic diseases. Mutations in the human DCX gene result in abnormal neuronal migration, epilepsy, and mental retardation; mutations in RPI are associated with a form of inherited blindness, and DCDC2 has been associated with dyslectic reading disabilities. Results: The DCX-repeat gene family is composed of eleven paralogs in human and in mouse. Its evolution was followed across vertebrates, invertebrates, and was traced to unicellular organisms, thus enabling following evolutionary additions and losses of genes or domains. The N-terminal and C-terminal DCX domains have undergone sub-specialization and divergence. Developmental in situ hybridization data for nine genes was generated. In addition, a novel co-expression analysis for most human and mouse DCX superfamily-genes was performed using high-throughput expression data extracted from Unigene. We performed an in-depth study of a complete gene superfamily using several complimentary methods. Conclusion: This study reveals the existence and conservation of multiple members of the DCX superfamily in different species. Sequence analysis combined with expression analysis is likely to be a useful tool to predict correlations between human disease and mouse models. The sub-specialization of some members due to restricted expression patterns and sequence divergence may explain the successful addition of genes to this family throughout evolution.
-
(2006) Molecular and Cellular Neuroscience. 32, 1-2, p. 15-26 Abstract
Mutations in doublecortin (DCX) cause X-linked lissencephaly ("smooth brain") and double cortex syndrome in humans. DCX is highly phosphorylated in migrating neurons. Here, we demonstrate that dephosphorylation of specific sites phosphorylated by JNK is mediated by Neurabin II, which recruits the phosphatase PP1. During cortical development, the expression pattern of PP1 is widespread, while the expression of DCX and Neurabin II is dynamic, and they are coexpressed in migrating neurons. In vitro, DCX is site-specific dephosphorylated by PP1 without the presence of Neurabin II, this dephosphorylation requires an intact RVXF motif in DCX. Overexpression of the coiled-coil domain of Neurabin II, which is sufficient for interacting with DCX and recruiting the endogenous Neurabin II with PP1, induced dephosphorylation of DCX on one of the JNK-phosphorylated sites. We hypothesize that the transient recruitment of DCX to different scaffold proteins, JIP-1/2, which will regulate its phosphorylation by JNK, and Neurabin II, which will regulate its dephosphorylation by PP1, plays an important role in normal neuronal migration.
-
(2006) Cell Cycle. 5, 9, p. 976-983 Abstract
The doublecortin-like (DCX) domains serve as protein-interaction platforms. DCX tandem domains appear in the product of the X-linked doublecortin (DCX) gene, in retinitis pigmentosa-1 (RP1), as well as in other gene products. Mutations in the human DCX gene are associated with abnormal neuronal migration, epilepsy, and mental retardation; mutations in RP1 are associated with a form of inherited blindness, while DCDC2 has been associated with dyslectic reading disabilities. Motivated by the possible importance of this gene family, a thorough analysis to detect all family members in the mouse was conducted. The DCX-repeat gene superfamily is composed of eleven paralogs, and we cloned the DCX domains from nine different genes. Our study questioned which functions attributed to the DCX domain, are conserved among the different members. Our results suggest that the proteins with the DCX-domain have conserved and unique roles in microtubule regulation and signal transduction. All the tested proteins stimulated microtubule assembly in vitro. Proteins with tandem repeats stabilized the microtubule cytoskeleton in transfected cells, while those with single repeats localized to actin-rich subcellular structures, or the nucleus. All tested proteins interacted with components of the JNK/MAP-kinase pathway, while only a subset interacted with Neurabin 2, and a nonoverlapping group demonstrated actin association. The sub-specialization of some members due to confined intracellular localization, and protein interactions may explain the success of this superfamily.
-
(2006) Nature Cell Biology. 8, 1, p. 11-13 Abstract
p27kip1, once confined to control of the cell cycle, has more recently been identified as a key regulator of cell migration. In neuronal cells, this function of p27kip1 is regulated by Cdk5.
-
(2006) Neuroscience. 139, 4, p. 1289-1300 Abstract
Both neural development and prefrontal cortex function are known to be abnormal in schizophrenia and bipolar disorder. In order to test the hypothesis that these features may be related with genes that regulate neuronal migration, we analyzed two genomic regions: the lissencephaly critical region (chromosome 17p) encompassing the LIS1 gene and which is involved in human lissencephaly; and the genes related to the platelet-activating-factor, functionally related to LIS1, in 52 schizophrenic patients, 36 bipolar I patients and 65 normal control subjects. In addition, all patients and the 25 control subjects completed a neuropsychological battery. Thirteen (14.8%) patients showed genetic variations in either two markers related with lissencephaly or in the platelet-activating-factor receptor gene. These patients performed significantly worse in the Wisconsin Card Sorting Test-Perseverative Errors in comparison with patients with no lissencephaly critical region/platelet-activating-factor receptor variations. The presence of lissencephaly critical region/platelet-activating-factor receptor variations was parametrically related to perseverative errors, and this accounted for 17% of the variance (P=0.0001). Finally, logistic regression showed that poor Wisconsin Card Sorting Test-Perseverative Errors performance was the only predictor of belonging to the positive lissencephaly critical region/platelet-activating-factor receptor group. These preliminary findings suggest that the variations in genes involved in neuronal migration predict the severity of the prefrontal cognitive deficits in both disorders.
-
(2006) Neuron. 49, 1, p. 25-39 Abstract
The mechanisms controlling neurogenesis during brain development remain relatively unknown. Through a differential protein screen with developmental versus mature neural tissues, we identified a group of developmentally enriched microtubule-associated proteins (MAPs) including doublecortin-like kinase (DCLK), a protein that shares high homology with doublecortin (DCX). DCLK, but not DCX, is highly expressed in regions of active neurogenesis in the neocortex and cerebellum. Through a dynein-dependent mechanism, DCLK regulates the formation of bipolar mitotic spindles and the proper transition from prometaphase to metaphase during mitosis. In cultured cortical neural progenitors, DCLK RNAi Lentivirus disrupts the structure of mitotic spindles and the progression of M phase, causing an increase of cell-cycle exit index and an ectopic commitment to a neuronal fate. Furthermore, both DCLK gain and loss of function in vivo specifically promote a neuronal identity in neural progenitors. These data provide evidence that DCLK controls mitotic division by regulating spindle formation and also determines the fate of neural progenitors during cortical neurogenesis.
2005
-
(2005) Cell Cycle. 4, 11, p. 1632-1640 Abstract
The presence of a conserved protein motif usually implies common functional features. Here, we focused on the LisH (LIS1 homology) domain, which is found in multiple proteins, and have focused on three involved in human genetic diseases; LIS1, Transducin β-like 1X (TBL1) and Oral-facial-digital type 1 (OFD1). The recently solved structure of the LisH domain in the N-terminal region of LIS1 depicted it as a novel dimerization motif. Our findings indicated that the LisH domain of both LIS1 and TBL1 is essential for in vitro oligomerization. Furthermore, our study disclosed novel in vivo features of the LisH motif. Mutations in conserved LisH amino acids significantly reduced both the protein half-life of LIS1, TBL1, and OFD1, and dramatically affected specific intracellular localizations of these proteins. LIS1 mutated in the LisH domain induced its localization to the actin filaments. TBL1 mutated in the LisH domain was not imported into the nucleus. Mutations in OFD1 modified its localization to the Golgi apparatus and in some cases also to the nucleus. In summary, the LisH domain may participate in protein dimerization, affect protein half-life, and may influence specific cellular localizations. Our results allow the prediction that mutations within the LisH motif are likely to result in pathogenic consequences in genes associated with genetic diseases.
-
Binding of microtubule-associated protein 1B to LIS1 affects the interaction between dynein and LIS1(2005) Biochemical Journal. 389, 2, p. 333-341 Abstract
For neuronal migration to occur, the cell must undergo morphological changes that require modifications of the cytoskeleton. Several different MAPs (microtubule-associated proteins) or actin-binding proteins are proposed to be involved in the migration of neurons. Therefore we have specifically analysed how two members of the MAP family, MAP1B and LIS1 (lissencephaly-related protein 1), interact with one another and participate in neuronal migration. Our results indicate that, in hippocampal neurons, MAP1B and LIS1 co-localize, associate and interact with each another. The interaction between these two MAPs is regulated by the phosphorylation of MAP1B. Furthermore, this interaction interferes with the association between LIS1 and the microtubule-dependent molecular motor, dynein. Clearly, the differential binding of these cytoskeletal proteins could regulate the functions attributed to the LIS1-dynein complex, including those related to extension of the neural processes necessary for neuronal migration.
-
(2005) Retrovirology. 2, 6. Abstract
Background: HIV-1 Tat activates transcription of HIV-1 viral genes by inducing phosphorylation of the C-terminal domain (CTD) of RNA polymerase II (RNAPII). Tat can also disturb cellular metabolism by inhibiting proliferation of antigen-specific T lymphocytes and by inducing cellular apoptosis. Tat-induced apoptosis of T-cells is attributed, in part, to the distortion of microtubules polymerization. LIS1 is a microtubule-associated protein that facilitates microtubule polymerization. Results: We identified here LIS1 as a Tat-interacting protein during extensive biochemical fractionation of T-cell extracts. We found several proteins to co-purify with a Tat-associated RNAPII CTD kinase activity including LIS1, CDK7, cyclin H, and MAT1. Tat interacted with LIS1 but not with CDK7, cyclin H or MAT1 in vitro. LIS1 also co-immunoprecipitated with Tat expressed in HeLa cells. Further, LIS1 interacted with Tat in a yeast two-hybrid system. Conclusion: Our results indicate that Tat interacts with LIS1 in vitro and in vivo and that this interaction might contribute to the effect of Tat on microtubule formation.
-
(2005) Molecular Neurobiology. 31, 1-3, p. 117-134 Abstract
One of the key features in development is the reutilization of successful signaling pathways. Here, we emphasize the involvement of the Wnt pathway, one of the five kinds of signal transduction pathway predominating early embryonic development of all animals, in regulating the formation of brain structure. We discuss the interrelationships between the Wnt and reelin pathways in the regulation of cortical layering. We summarize data emphasizing key molecules, which, when mutated, result in abnormal brain development. This integrated view, which is based on conservation of pathways, reveals the relative position of participants in the pathway, points to control mechanisms, and allows raising testable working hypotheses. Nevertheless, although signaling pathways are highly conserved from flies to humans, the overall morphology is not. We propose that future studies directed at understanding of diversification will provide fruitful insights on mammalian brain formation.
-
(2005) Cellular and Molecular Life Sciences. 62, 4, p. 425-434 Abstract
Proper human brain formation is dependent upon the integrated activity of multiple genes. Malfunctioning of key proteins results in brain developmental abnormalities. Mutation(s) in the LIS1 gene or the X-linked gene doublecortin (DCX) results in a spectrum of disorders including lissencephaly, or 'smooth brain', and subcortical band heterotopia, or 'doublecortex'. Here, we will focus on a particular subset of missense mutations in these two genes and their effect on protein structure and function.
2004
-
(2004) Cell Cycle. 3, 6, p. 745-749 Abstract
The mammalian cortex is generally subdivided into six organized layers, which are formed during development in an organized fashion. This organized cortical layering is disrupted in case of mutations in the doublecortin (DCX) gene. DCX is a Microtubule Associated Protein (MAP). However, besides stabilization of microtubules, it may be involved in additional functions. The participation of this molecule in signal transduction is beginning to emerge via discovery of interacting molecules and its regulation by phosphorylation using several different kinases. We raise the hypothesis, that the combinatorial phosphorylation of DCX by different kinases and at different, sites may be a molecular regulatory switch in the transition of a migrating neuron through multiple phases of migration. Our recent research has suggested the involvement of DCX in the JNK (Jun-N-terminal Kinase) pathway. The JNK pathway is linked to the reelin pathway, known to regulate cortical layering. Positioning of DCX in this signaling pathway opens up additional possibilities of understanding how migrating neurons are controlled.
-
(2004) Structure. 12, 6, p. 987-998 Abstract
Mutations in the Lis1 gene result in lissencephaly (smooth brain), a debilitating developmental syndrome caused by the impaired ability of postmitotic neurons to migrate to their correct destination in the cerebral cortex. Sequence similarities suggest that the LIS1 protein contains a C-terminal seven-blade β-propeller domain, while the structure of the N-terminal fragment includes the LisH (Lis-homology) motif, a pattern found in over 100 eukaryotic proteins with a hitherto unknown function. We present the 1.75 Å resolution crystal structure of the N-terminal domain of mouse LIS1, and we show that the LisH motif is a novel, thermodynamically very stable dimerization domain. The structure explains the molecular basis of a low severity form of lissencephaly.
-
(2004) EMBO Journal. 23, 4, p. 823-832 Abstract
Mutations in the X-linked gene DCX result in lissencephaly in males, and abnormal neuronal positioning in females, suggesting a role for this gene product during neuronal migration. In spite of several known protein interactions, the involvement of DCX in a signaling pathway is still elusive. Here we demonstrate that DCX is a substrate of JNK and interacts with both c-Jun N-terminal kinase (JNK) and JNK interacting protein (JIP). The localization of this signaling module in the developing brain suggests its functionality in migrating neurons. The localization of DCX at neurite tips is determined by its interaction with JIP and by the interaction of the latter with kinesin. DCX is phosphorylated by JNK in growth cones. DCX mutated in sites phosphorylated by JNK affected neurite outgrowth, and the velocity and relative pause time of migrating neurons. We hypothesize that during neuronal migration, there is a need to regulate molecular motors that are working in the cell in opposite directions: kinesin (a plus-end directed molecular motor) versus dynein (a minus-end directed molecular motor).
2003
-
(2003) Molecular Reproduction and Development. 66, 2, p. 134-142 Abstract
Homozygous deletion of the Lis1 gene (Lis1-/-) in mouse resulted in early embryonic lethality immediately after embryo implantation by an undefined mechanism. We seek to define the nature of this demise. LIS1 (pafah1b1) is a 46 kDa protein with seven tryptophan-aspartate (WD) repeats. It docks with many proteins and has been implicated in microtubular function, cell division, intercellular transport, and nuclear and cellular motility. Combined Western and quantitative reverse transcriptase-polymerase chain reaction (RT-PCR) analyses showed that LIS1 expression from the blastocyst stage required new transcription from the embryonic genome. Consequently, the death of post-implantation embryos may not reflect the first time during development that LIS1 was required, rather, it may reflect the first time following depletion of gametic stores that its actions were essential. Following culture of blastocysts in vitro for 96 hr the inner cell mass (ICM) of null embryos were significantly smaller than ICM of wild-type siblings. Normal blastocyst outgrowths after 96-hr culture had high levels of LIS1 expression in the outer cells of developing ICM and extensive expression in trophoblast cells. Lis1 -/- embryos had significantly smaller trophoblast nuclei than wild-type embryos. The results show that LIS1 expression is required for the continued normal development of the ICM and optimal trophoblast giant cell formation.
-
(2003) Journal of Biological Chemistry. 278, 40, p. 38740-38748 Abstract
Mutations in one allele of the human LIS1 gene cause a severe brain malformation, lissencephaly. Although most LIS1 mutations involve deletions, several point mutations with a single amino acid alteration were described. Patients carrying these mutations reveal variable phenotypic manifestations. We have analyzed the functional importance of these point mutations by examining protein stability, folding, intracellular localization, and protein-protein interactions. Our data suggest that the mutated proteins were affected at different levels, and no single assay could be used to predict the lissencephaly phenotype. Most interesting are those mutant proteins that retain partial folding and interactions. In the case of LIS1 mutated in F31S, the cellular phenotype may be modified by overexpression of specific interacting proteins. Overexpression of the PAF-AH α1 subunit dissolved aggregates induced by this mutant protein and increased its half-life. Overexpression of NudE or NudEL localized this mutant protein to spindle poles and kinetochores but had no effect on protein stability. Our results implicate that there are probably different biochemical and cellular mechanisms obstructed in each patient yielding the varied lissencephaly phenotypes.
2002
-
(2002) Nature Genetics. 32, 3, p. 341-342 Abstract
Research on homeobox genes has shown them to have crucial roles in many developmental processes. A new study on the homeobox transcription factor, ARX, offers insights into neuronal migration and, surprisingly, testis development.
-
(2002) Journal of Biological Chemistry. 277, 20, p. 17696-17705 Abstract
Alternative splicing of mRNA transcripts expands the range of protein products from a single gene locus. Several splice variants of DCLK (doublecortin-like kinase) have previously been reported. Here, we report the genomic organization underlying the splice variants of DCLK and examine the expression profile of two splice variants affecting the kinase domain of DCLK and CPG16 (candidate plasticity gene 16), one containing an Arg-rich domain and the other affecting the C terminus of the protein. These splice alternatives were differentially expressed in embryonic and adult brain. Both splice variants disrupted DCLK PEST domains; however, all splice variants remained sensitive to proteolysis by calpain. The adult-specific C-terminal splice variant of DCLK had reduced autophosphorylation activity, but similar kinase activity for myelin basic protein relative to the embryonic splice variant. The splice variant adding an Arg-rich domain gained an autophosphorylation site at Ser-382. Although this protein isoform was expressed mainly in the adult brain, the phosphorylated form was strongly enriched in embryonic brain and adult olfactory bulb, suggesting a possible role in migrating neurons.
-
(2002) Molecular Psychiatry. 7, 1, p. 12-16 Abstract
LIS1 is one of the genes that has a principle role in brain development since hemizygote mutations in LIS1 result in a severe brain malformation known as lissencephaly ('smooth brain'). LIS1 is a WD repeat protein and is known to be involved in several protein complexes that are likely to play a functional role in brain development. We discuss here the brain developmental phenotype observed in mice heterozygote for an N-terminal truncated LIS1 protein in view of known LIS1 protein interactions.
-
(2002) Molecular and Cellular Biology. 22, 9, p. 3089-3102 Abstract
CLIP-170 is a plus-end tracking protein which may act as an anticatastrophe factor. It has been proposed to mediate the association of dynein/dynactin to microtubule (MT) plus ends, and it also binds to kinetochores in a dynein/dynactin-dependent fashion, both via its C-terminal domain. This domain contains two zinc finger motifs (proximal and distal), which are hypothesized to mediate protein-protein interactions. LIS1, a protein implicated in brain development, acts in several processes mediated by the dynein/dynactin pathway by interacting with dynein and other proteins. Here we demonstrate colocalization and direct interaction between CLIP-170 and LIS1. In mammalian cells, LIS1 recruitment to kinetochores is dynein/dynactin dependent, and recruitment there of CLIP-170 is dependent on its site of binding to LIS1, located in the distal zinc finger motif. Overexpression of CLIP-170 results in a zinc finger-dependent localization of a phospho-LIS1 isoform and dynactin to MT bundles, raising the possibility that CLIP-170 and LIS1 regulate dynein/dynactin binding to MTs. This work suggests that LIS1 is a regulated adapter between CLIP-170 and cytoplasmic dynein at sites involved in cargo-MT loading, and/or in the control of MT dynamics.
-
(2002) Molecular Psychiatry. 7, 1, p. 12-16 Abstract
LIS1 is one of the genes that has a principle role in brain development since hemizygote mutations in LIS1 result in a severe brain malformation known as lissencephaly ('smooth brain'). LIS1 is a WD repeat protein and is known to be involved in several protein complexes that are likely to play a functional role in brain development. We discuss here the brain developmental phenotype observed in mice heterozygote for an N-terminal truncated LIS1 protein in view of known LIS1 protein interactions.
2001
-
(2001) Journal of Biological Chemistry. 276, 39, p. 36397-36403 Abstract
Doublecortin-like kinase (DCLK) is widely expressed in postmitotic neurons throughout the embryonic nervous system. DCLK consists of an N-terminal doublecortin domain, responsible for its localization to microtubules, and a C-terminal serine-threonine kinase domain. Here we report that DCLK is a physiological substrate for the cysteine protease calpain. Cleavage of DCLK by calpain severs the kinase domain from its microtubule anchorage domain and releases it into the cytoplasm. The isolated kinase domain retains catalytic activity and is structurally similar to CPG16, a second product of the DCLK gene expressed in the adult brain that lacks the doublecortin domain. We propose that in neurons cleavage of DCLK by calpain represents a calcium-responsive mechanism to regulate localization of the DCLK kinase domain.
-
(2001) Proceedings of the National Academy of Sciences of the United States of America. 98, 11, p. 6429-6434 Abstract
Lissencephaly is a severe brain malformation in humans. To study the function of the gene mutated in lissencephaly (LIS1), we deleted the first coding exon from the mouse Lis1 gene. The deletion resulted in a shorter protein (sLIS1) that initiates from the second methionine, a unique situation because most LIS1 mutations result in a null allele. This mutation mimics a mutation described in one lissencephaly patient with a milder phenotype. Homozygotes are early lethal, although heterozygotes are viable and fertile. Most strikingly, the morphology of cortical neurons and radial glia is aberrant in the developing cortex, and the neurons migrate more slowly. This is the first demonstration, to our knowledge, of a cellular abnormality in the migrating neurons after Lis1 mutation. Moreover, cortical plate splitting and thalamocortical innervation are also abnormal. Biochemically, the mutant protein is not capable of dimerization, and enzymatic activity is elevated in the embryos, thus a demonstration of the in vivo role of LIS1 as a subunit of PAF-AH. This mutation allows us to determine a hierarchy of functions that are sensitive to LIS1 dosage, thus promoting our understanding of the role of LIS1 in the developing cortex.
-
(2001) Human Molecular Genetics. 10, 10, p. 1061-1070 Abstract
Mutations in doublecortin (DCX) result in X-linked lissencephaly in males. To explore the role of DCX in differentiation and signal transduction we over-expressed DCX in PC12 cells. Our results indicate that DCX stabilizes microtubules and inhibits neurite outgrowth in nerve growth factor-induced differentiation. However, neurite length is increased when differentiation is induced by epidermal growth factor and forskolin or by dibutyryl-cAMP. Furthermore, CREB-mediated transcription is downregulated, supporting the notion that cytoskeletal regulatory proteins can affect the transcriptional state of a cell. Using different constructs and mutations we reach the conclusion that microtubule stabilization is a key factor, but not the only one, in controlling neurite extension. Overexpression of a mutation found in a lissencephaly patient (S47R), completely blocks neurite outgrowth. We propose that these functions are important during normal and abnormal brain development.
2000
-
(2000) Human Molecular Genetics. 9, 15, p. 2205-2213 Abstract
Mutations in either LIS1 or DCX are the most common cause for type I lissencephaly. Here we report that LIS1 and DCX interact physically both in vitro and in vivo. Epitope-tagged DCX transiently expressed in COS cells can be co-immunoprecipitated with endogenous LIS1. Furthermore, endogenous DCX could be co-immunoprecipitated with endogenous LIS1 in embryonic brain extracts, demonstrating an in vivo association. The two protein products also co-localize in transfected cells and in primary neuronal cells. In addition, we demonstrate homodimerization of DCX in vitro. Using fragments of both LIS1 and DCX, the domains of interaction were mapped. LIS1 and DCX interact with tubulin and microtubules. Our results suggest that addition of DCX and LIS1 to tubulin enhances polymerization in an additive fashion. In in vitro competition assays, when LIS1 is added first, DCX competes with LIS1 in its binding to microtubules, but when DCX is added prior to the addition of LIS1 it enhances the binding of LIS1 to microtubules. We conclude that LIS1 and DCX cross-talk is important to microtubule function in the developing cerebral cortex.
-
(2000) European Neuropsychopharmacology. 10, 5, p. 389-395 Abstract
The hippocampus, a medial temporal lobe structure, is often considered to play an important role in the pathophysiology of schizophrenia. Recent developments of neuroimaging and molecular postmortem techniques have significantly increased our ability to study the role of discrete brain regions in the pathophysiology of schizophrenia. This article describes animal models, structural, histological, molecular biology, and neuropsychological evidence for the involvement of the hippocampus in the pathophysiology of schizophrenia. The major findings in schizophrenic patients are decreased volumes, hypometabolism, and cytoarchitectural abnormalities which are more robust on the left hippocampus, as well as verbal memory impairment. It is yet to be determined whether these changes are neurodevelopmental or neurodegenerative in nature. Overall, these findings indicate that there are subtle changes in the hippocampus of schizophrenic patients. More comprehensive and focused hippocampal research in schizophrenia is required to elucidate the contribution of this intriguing brain structure to the pathophysiology of schizophrenia. (C) 2000 Elsevier Science B.V.
-
(2000) Proteins-Structure Function And Genetics. 39, 1, p. 1-8 Abstract
The mammalian intracellular brain platelet-activating factor acetylhydrolase, implicated in the development of cerebral cortex, is a member of the phospholipase A2 superfamily. It is made up of a homodimer of the 45 kDa LIS1 protein (a product of the causative gene for type I lissencephaly) and a pair of homologous 26-kDa α-subunits which account for all the catalytic activity. LIS1 is hypothesized to regulate nuclear movement in migrating neurons through interactions with the cytoskeleton, while the α-subunits, whose structure is known, contain a trypsin-like triad within the framework of a unique tertiary fold. The physiological significance of the association of the two types of subunits is not known. In an effort to better understand the function of the complex we turned to genomic data mining in search of related proteins in lower eukaryotes. We found that the Drosophila melanogaster genome contains homologs of both a- and β-subunits, and we cloned both genes. The α-subunit homolog has been overexpressed, purified and crystallized. It lacks two of the three active-site residues and, consequently, is catalytically inactive against PAF-AH (Ib) substrates. Our study shows that the β-subunit homolog is highly conserved from Drosophila to mammals and is able to interact with the mammalian α-subunits but is unable to interact with the Drosophila α-subunit. (C) 2000 Wiley- Liss, Inc.
-
(2000) Human Molecular Genetics. 9, 5, p. 703-712 Abstract
Mutations in the X-linked gene doublecortin (DCX) result in lissencephaly in males or subcortical laminar heterotopia ('double cortex') in females. Various types of mutation were identified and the sequence differences included nonsense, splice site and missense mutations throughout the gene. Recently, we and others have demonstrated that DCX interacts and stabilizes microtubules. Here, we performed a detailed sequence analysis of DCX and DCX-like proteins from various organisms and defined an evolutionarily conserved Doublecortin (DC) domain. The domain typically appears in the N-terminus of proteins and consists of two tandemly repeated 80 amino acid regions. In the large majority of patients, missense mutations in DCX fall within the conserved regions. We hypothesized that these repeats may be important for microtubule binding. We expressed DCX or DCLK (KIAA0369) repeats in vitro and in vivo. Our results suggest that the first repeat binds tubulin but not microtubules and enhances microtubule polymerization. To study the functional consequences of DCX mutations, we overexpressed seven of the reported mutations in COS7 cells and examined their effect on the microtubule cytoskeleton. The results demonstrate that some of the mutations disrupt microtubules. The most severe effect was observed with a tyrosine to histidine mutation at amino acid 125 (Y125H). Produced as a recombinant protein, this mutation disrupts microtubules in vitro at high molar concentration. The positions of the different mutations are discussed according to the evolutionarily defined DC-repeat motif. The results from this study emphasize the importance of DCX-microtubule interaction during normal and abnormal brain development.
-
-
(2000) Development Genes and Evolution. 210, 1, p. 51-54 Abstract
We have isolated the chicken LIS1 homolog, chLIS1, with DNA sequence similarity of over 68% to the human cDNA and 99% amino acid identity. Additionally, we describe the pattern of chLIS1 expression in the chicken embryo. The early embryonic expression is highly specific to the developing nervous system, whereas later the expression is more widespread.
-
(2000) Molecular and Cellular Neuroscience. 16, 5, p. 529-541 Abstract
Doublecortin-like kinase (DCLK) shares sequence similarity to Doublecortin (DCX) in its N-terminal region. It contains the evolutionary conserved DC repeat motif as well a C-terminal kinase domain. Ectopic expression of DCLK in COS cells results in colocalization with microtubules, and phosphorylated DCLK copurifies with microtubules during assembly from embryonic brain extract. During brain development DCLK is expressed mainly in postmigratory neurons in a similar pattern to DCX. We demonstrate that DCLK is a microtubule-associated active protein kinase expressed in growth cones of postmitotic neurons.
1999
-
(1999) European Journal of Biochemistry. 266, 3, p. 1011-1020 Abstract
Mutations in the LIS1 gene may result in severe abnormalities of brain cortical layering known as lissencephaly. Most lissencephaly-causing LIS1 mutations are deletions that encompass the entire gene, therefore the mechanism of the disease is regarded as haploinsufficiency. So far, 13 different intragenic mutations have been reported: one point mutation, H149R; deletion of exon 9, which results in deleted acids A301-334; deletion of exon 4, which results in deleted amino acids A40-64;10 mutations resulting in truncated proteins and one predicted to result in extra amino acids. We studied the consequences of the point mutation, deletion mutation and one of the reported truncations. In order to study LIS 1 structure function, We introduced an additional point mutation and other truncations in different regions of the protein. The consequences of these mutations to protein folding were studied by gel filtration, sucrose density gradient centrifugation and measuring resistance to trypsin cleavage. On the basis of our results, we suggest that all truncation mutations and lissencephaly- causing point mutations or internal deletion result in a reduction in the amount of correctly folded LIS1 protein.
-
(1999) Journal of Neuroscience Research. 58, 4, p. 567-575 Abstract
During embryonic development, the cerebral cortex attains its characteristic adult laminated structure. The finding that X-linked lissencephaly patients harbor mutations in the doublecortin gene implicated this gene product in the process of corticogenesis. An autosomal human gene, KIAA0369, with a high level of similarity to doublecortin, has been cloned from human adult brain. This gene product contains a kinase domain in addition to a doublecortin-like domain. In order to evaluate whether this doublecortin-like kinase also plays a role during brain development, we cloned and studied the expression pattern of the mouse homolog. Three cDNA products of this gene were cloned: one, doublecortin-like kinase, the second containing only the doublecortin-like region, and the third containing only the kinase domain, a homolog of the previously cloned rat CPG16 gene. We studied doublecortin-like kinase expression in mouse using Northern blot analysis, in situ hybridization, and Western blot analysis, and conclude that doublecortin-like kinase is expressed in multiple regions of embryonic brain including the developing cerebral cortex.
-
(1999) European Journal of Biochemistry. 265, 1, p. 181-188 Abstract
Lissencephaly, a severe brain malformation, may be caused by mutations in the LIS1 gene. LIS1 encodes a microtubule-associated protein (MAP) that is also part of the enzyme complex, platelet-activating factor acetylhydrolase. LIS1 is also found in a complex with two protein kinases; a T-cell Tat- associated kinase, which contains casein-dependant kinase (CDK) activating kinase (CAK), as well as CAK-inducing activity, and with a spleen protein- tyrosine kinase similar to the catalytic domain of p72syk. As phosphorylation is one of the ways to control cellular localization and protein-protein interactions, we investigated whether LIS1 undergoes this posttranslational modification. Our results demonstrate that LIS1 is a developmentally regulated phosphoprotein. Phosphorylated LIS1 is mainly found in the MAP fraction. Phosphoamino acid analysis revealed that LIS1 is phosphorylated on serine residues. Alkaline phosphatase treatment reduced the number of visible LIS1 isoforms. In-gel assays demonstrate a 50-kDa LIS1 kinase that is enriched in microtubule-associated fractions. In vitro, LIS1 was phosphorylated by protein kinase CKII (casein kinase II), but not many other kinases that were tested. We suggest that LIS1 activity may be regulated by phosphorylation.
-
(1999) Journal of Neuroscience Research. 57, 2, p. 176-184 Abstract
Lissencephaly patients are born with severe brain malformations and suffer from recurrent seizures. LIS1, the gene mutated in isolated lissencephaly patients, is a subunit of the heterotrimeric cytosolic enzyme platelet-activating factor acetylhydrolase (PAF-AH), interacts with tubulin, and affects microtubule dynamics. In order to gain molecular insights into the possible involvement of LIS1 in seizures in lissencephaly patients, we induced seizures in rats by injection of kainate. PAF-AH activity was markedly reduced as early as 30 min following initiation of seizures, making this parameter a sensitive indicator of seizure events. PAF-AH activity returned to and surpassed control values I week following initiation of seizures. Expression of LIS1 in the dentate gyrus changed significantly in a manner similar to that of PAF-AH enzymatic activity. This is the first correlation found between LIS1 expression and PAF-AH activity. Furthermore, the expression of the α2 catalytic subunit, which is the major PAF-AH catalytic subunit in rat adult brain, changed in a dramatic fashion. An additional higher-mobility LIS1 crossreactive band was detected in samples isolated a week following seizure occurrence. This LIS1 isoform was enriched in the microtubule-associated fraction. We propose that LIS1 expression is an important factor in regulation of PAF-AH activity. We postulate that reductions in LIS1 protein levels found in lissencephaly patients may render them more susceptible to seizures.
-
(1999) Neuron. 23, 2, p. 247-256 Abstract
Recently, we and others reported that the doublecortin gene is responsible for X-linked lissencephaly and subcortical laminar heterotopia. Here, we show that Doublecortin is expressed in the brain throughout the period of corticogenesis in migrating and differentiating neurons. Immunohistochemical studies show its localization in the soma and leading processes of tangentially migrating neurons, and a strong axonal labeling is observed in differentiating neurons. In cultured neurons, Doublecortin expression is highest in the distal parts of developing processes. We demonstrate by sedimentation and microscopy studies that Doublecortin is associated with microtubules (MTs) and postulate that it is a novel MAP. Our data suggest that the cortical dysgeneses associated with the loss of Doublecortin function might result from abnormal cytoskeletal dynamics in neuronal cell development.
-
(1999) FEBS Letters. 451, 2, p. 99-102 Abstract
Platelet-activating factor is a phospholipid with several documented roles in the pre-implantation embryo. Enzymes that belong to the platelet-activating factor acetylhydrolases family inactivate platelet-activating factor. Cytosolic platelet-activating factor acetylhydrolase (Ib) is a heterotetramer composed of two catalytic subunits (α1/α2) and two regulatory LIS1 subunits. The expression of these components was monitored in the mouse oocytes and zygotes using reverse-transcribed PCR and Western blot analysis. Interestingly, these proteins are expressed in the oocyte and zygote and their expression increases after fertilization, probably due to stabilization of maternal RNA. Lis1 mRNA transcription also increases after fertilization. However, assaying for expression of a specific paternal LIS1 isoform detected no zygotic translation in the one cell stage. These findings suggest a potential role for platelet-activating factor acetylhydrolase (Ib) components in the early mouse embryo. Copyright (C) 1999 Federation of European Biochemical Societies.
-
(1999) Human Molecular Genetics. 8, 9, p. 1599-1610 Abstract
X-linked lissencephaly is a severe brain malformation affecting males. Recently it has been demonstrated that the doublecortin gene is implicated in this disorder. In order to study the function of Doublecortin, we analyzed the protein upon transfection of COS cells. Doublecortin was found to bind to the microtubule cytoskeleton. In vitro assays (using biochemical methods, DIC microscopy and electron microscopy) demonstrate that Doublecortin binds microtubules directly, stabilizes them and causes bundling. In vivo assays also show that Doublecortin stabilizes microtubules and causes bundling. Doublecortin is a basic protein with an isoelectric point of 10, typical of microtubule-binding proteins. However, its sequence contains no known microtubule-binding domain(s). The results obtained in this study with Doublecortin and our previous work on another lissencephaly gene (LIS1) emphasize the central role of regulation of microtubule dynamics and stability during neuronal morphogenesis.
-
(1999) Molecular Neurobiology. 20, 2-3, p. 143-156 Abstract
Formation of our highly structured human brain involves a cascade of events, including differentiation, fate determination, and migration of neural precursors. In humans, unlike many other organisms, the cerebral cortex is the largest component of the brain. As in other mammals, the human cerebral cortex is located on the surface of the telencephalon and generally consists of six layers that are formed in an orderly fashion. During neuronal development, newly born neurons, moving in a radial direction, must migrate through previously formed layers to reach their proper cortical position. This is one of several neuronal migration routes that takes place in the developing brain; other modes of migration are tangential. Abnormal neuronal migration may in turn result in abnormal development of the cortical layers and deleterious consequences, such as Lissencephaly. Lissencephaly, a severe brain malformation, can be caused by mutations in one of two known genes: LIS1 and doublecortin (DCX). Recent in vitro and in vivo studies, report on possible functions for these gene products.
1998
-
(1998) International Journal of Molecular Medicine. 1, 5, p. 849-853 Abstract
Lissencephaly is a relatively common brain malformation. Lissencephaly type 1 is characterized by the smooth appearance of the cortex and the presence of four abnormally positioned layers instead of the normal six. Lissencephaly is considered to be an abnormality in neuronal migration. The gene mutated in type 1 lissencephaly was cloned by us and designated LIS1. Recently, several genes involved in cortical development have been cloned in the mouse. In human an additional X-linked lissencephaly gene has been identified. We summarize here our current knowledge on the LIS1 gene and its function. It has been identified as a non-catalytic subunit of PAF-acetylhydrolase, a heterotrimeric enzyme which inactivates the platelet-activating factor (PAF). In addition, we have demonstrated that LIS1 interacts with tubulin, and affects the dynamics properties of microtubles. LIS1 contains seven WD repeats and may structurally resemble the beta-subunit of heterotrimeric G proteins. Interestingly, the catalytic subunit of PAF-acetylhydrolase was found to resemble the alpha subunit of heterotrimeric G proteins. We raise the possibility that LIS1 is part of an intracellular signaling pathway involved in neuronal migration.
-
(1998) Current Biology. 8, 10, p. 603-606 Abstract
Important clues to how the mammalian cerebral cortex develops are provided by the analysis of genetic diseases that cause cortical malformations [1-5]. People with Miller-Dieker syndrome (MDS) or isolated lissencephaly sequence (ILS) have a hemizygous deletion or mutation in the LIS1 gene [3,6]; both conditions are characterized by a smooth cerebral surface, a thickened cortex with four abnormal layers, and misplaced neurons [7,8]. LIS1 is highly expressed in the ventricular zone and the cortical plate [9,10], and its product, Lis1, has seven WD repeats [3]; several proteins with such repeats have been shown to interact with other polypeptides, giving rise to multiprotein complexes [11]. Lis1 copurifies with platelet-activating factor acetylhydrolase subunits α1 and α2 [12], and with tubulin; it also reduces microtubule catastrophe events in vitro [13]. We used a yeast two-hybrid screen to isolate new Lis1-interacting proteins and found a mammalian ortholog of NudC, a protein required for nuclear movement in Aspergillus nidulans [14]. The specificity of the mammalian NudC-Lis1 interaction was demonstrated by protein-protein interaction assays in vitro and by co-immunoprecipitation from mouse brain extracts. In addition, the murine mNudC and mLis1 genes are coexpressed in the ventricular zone of the forebrain and in the cortical plate. The interaction of Lis1 with NudC, in conjunction with the MDS and ILS phenotypes, raises the possibility that nuclear movement in the ventricular zone is tied to the specification of neuronal fates and thus to cortical architecture.
-
(1998) Journal of the American Academy of Child and Adolescent Psychiatry. 37, 2, p. 231-232 Abstract
1997
-
(1997) EMBO Journal. 16, 23, p. 6977-6984 Abstract
Forming the structure of the human brain involves extensive neuronal migration, a process dependent on cytoskeletal rearrangement. Neuronal migration is believed to be disrupted in patients exhibiting the developmental brain malformation lissencephaly. Previous studies have shown that LIS1, the defective gene found in patients with lissencephaly, is a subunit of the platelet-activating factor acetylhydrolase. Our results indicated that LIS1 has an additional function. By interacting with tubulin it suppresses microtubule dynamics. We detected LIS1 interaction with microtubules by immunostaining and co-assembly. LIS1-tubulin interactions were assayed by co-immunoprecipitation and by surface plasmon resonance changes. Microtubule dynamic measurements in vitro indicated that physiological concentrations of LIS1 indeed reduced microtubule catastrophe events, thereby resulting in a net increase in the maximum length of the microtubules. Furthermore, the LIS1 protein concentration in the brain, measured by quantitative Western blots, is high and is approximately one-fifth of the concentration of brain tubulin. Our new findings show that LIS1 is a protein exhibiting several cellular interactions, and the interaction with the cytoskeleton may prove to be the mode of transducing a signal generated by platelet-activating factor. We postulate that the LIS1-cytoskeletal interaction is important for neuronal migration, a process that is defective in lissencephaly patients.
1996
-
(1996) Biochemistry. 35, 44, p. 13985-13994 Abstract
The family of WD-repeat proteins comprises over 30 different proteins that share a highly conserved repeating motif [Neer, E. J., Schmidt, C. J., Numbudripad, R., and Smith, T. F. (1994) Nature 371, 297-300]. Members of this family include the signal-transducing G protein β subunit, as well as other proteins that regulate signal transduction, transcription, pre-mRNA splicing, cytoskeletal organization, and vesicular fusion. The crystal structure of one WD-repeat protein (Gβ) has now been solved (Wall et al., 1995; Sondek et al., 1996) and reveals that the seven repeating units form a circular, propeller-like structure with seven blades each made up of four β strands. It is very likely that all WD-repeat proteins form a similar structure. If so, it will be possible to use information about important surface regions of one family member to predict properties of another. If WD proteins form structures similar to Gβ, their hydrodynamic properties should be those of compact, globular proteins, and they should be resistant to cleavage by trypsin. However, the only studied example of a WD-repeat protein, Gβ, synthesized in vitro in a rabbit reticulocyte lysate, is unable to fold into a native structure without its partner protein Gγ. The non-WD- repeat amino terminal α helix of Gβ does not inhibit folding because Gβ does not fold even when this region is removed. It is not known whether all WD-repeat proteins are unable to fold when synthesized in an in vitro system. We synthesized seven members of the family in a rabbit reticulocyte lysate, determined their Stokes radius, sedimentation coefficient, and frictional ratio, and assayed their stability to trypsin. Our working definition of folding was that the proteins form globular, trypsin-resistant structures because, except for Gβγ, their functions are not known or cannot be assayed in reticulocyte lysates. We chose proteins that include amino and carboxyl extensions as well as proteins that are made up entirely of WD-repeats. We show that unlike Gβ, several proteins with WD-repeats are able to fold into globular proteins in a rabbit reticulocyte lysate. One protein, βTrcp, formed large aggregates like Gβ, suggesting that it may also require a partner protein. Despite the presence of many potential tryptic cleavage sites, all of the proteins that did fold gave stable large products on tryptic proteolysis, as predicted on the basis of the structure of Gβ. These studies suggest that other WD-repeat proteins are likely to form propeller structures similar to Gβ.
-
1995
-
(1995) Genomics. 30, 2, p. 251-256 Abstract
We report here the isolation of a novel cDNA, designatedLIS2, that maps to chromosome 2p11.2 byin situhybridization and demonstrates extremely high sequence similarity to the recently identifiedLIS1gene involved in Miller-Dieker lissencephaly at 17p13.3. Specific probes forLIS2revealed a pattern of expression resembling that ofLIS1, althoughLIS2is less abundant. Surprisingly, LIS2detected an additional, higher molecular weight transcript in adult skeletal muscle. Isolated YAC clones and P1 clones mapped byin situhybridization to two loci on chromosome 2, 2p11.2 and 2q13-q14. This hybridization was due to the existence ofLIS2pseudogeneLIS2Pon the long arm of chromosome 2.
-
(1995) Journal of Neuroscience. 15, 5 II, p. 3730-3738 Abstract
Miller-Dicker lissencephaly syndrome (MDS) is a human developmental brain malformation caused by neuronal migration defects resulting in abnormal layering of the cerebral cortex. LIS1, the gene defective in MDS, encodes a subunit of brain platelet-activating factor (PAF) acetylhydrolase which inactivates PAF, a neuroregulatory molecule. We have isolated murine cDNAs homologous to human LIS1 and mapped these to three different chromosomal loci (Lis1, Lis3, Lis4). The predicted sequences of murine Lis1 protein and its human homolog LIS1 are virtually identical. In the developing mouse and human, Lis1 and LIS1 genes were strongly expressed in the cortical plate. In the adult mouse Lis1 transcripts were abundant in cortex and hippocampus. The direct correlation between cortical defects in MDS patients and Lis1 expression in the murine cortex suggest that the mouse is a model system suitable to study the mechanistic basis of this intriguing genetic disease. Sequence data are deposited as L25108 for mouse Lis1 cDNA and L25109 for mouse Lis3-4 cDNA.
1994
-
REPORT OF THE 3RD INTERNATIONAL WORKSHOP ON HUMAN-CHROMOSOME-2 MAPPING 1994(1994) Cytogenetics and Cell Genetics. 67, 4, p. 216-237 Abstract
1989
-
Human sphingolipid activator protein-1 and sphingolipid activator protein-2 are encoded by the same gene.(1989) Journal of Molecular Neuroscience. 1, 4, p. 225-233 Abstract
Mixed oligonucleotide primers complementary to the translation product of the sphingolipid activator protein (SAP)-2 were used to generate a 144-base pair (bp) complementary DNA (cDNA). This cDNA probe was used to isolate a 2,649-nucleotide-long cDNA that was sequenced and found to contain coding sequences for two known activators of lysosomal enzymes, namely, the sphingolipid activator protein (SAP)-1 and SAP-2. The cDNA contains an open reading frame of 1,482 nucleotides and 1,167 nucleotides of 3'-nontranslated region, followed by a stretch of 24 residues of adenylic acid. At 20 nucleotides upstream from the poly(A) tail there is a consensus AATAAA polyadenylation signal that is preceded by another potential polyadenylation signal. The cDNA, designed SAP-1/SAP-2 cDNA, hybridizes with two human mRNA species of approximately 3 kb in length, which most probably arise from polyadenylation at different sites. There are higher amounts of steady-state RNA levels of the SAP-1/SAP-2 mRNA in skin fibroblasts in comparison to B cells. The steady-state SAP-1/SAP-2 mRNA levels in Gaucher B cells are higher than in their normal counterparts. There is one human SAP-1/SAP-2 gene that has been cloned and is localized on two approximately 5 kb BamHI fragments.
-
Characterization of mutations in gaucher patients by cDNA cloning(1989) American Journal of Human Genetics. 44, 3, p. 364-377 Abstract
Mutated cDNA clones containing the entire coding sequence of human glucocerebrosidase were isolated from libraries originated from Gaucher patients. Sequence analysis of a mutated cDNA derived from a type II Gaucher patient revealed a C-to-G transversion causing a substitution of an arginine for a proline at residue 415. This change creates a new cleavage site for the enzyme HhaI in the mutated cDNA. Allele-specific oligonucleotide hybridization made it possible to show that this mutation exists in the genomic DNA of the patient. From a cDNA library originated from a type I Gaucher patient, a mutated allele was cloned that contains a T-to-C transition causing a substitution of proline for leucine at residue 444 and creating a new NciI site. This mutation is identical to that described by S. Tsuji and colleagues in genomic DNA from type I, type II, and type III patients. Since the new NciI site generates RFLP, it was used to test the existence of this mutated allele in several Gaucher patients by Southern blot analysis. This allele was found in type I (Jewish and non-Jewish), type II, and type III Gaucher patients. These findings led us to conclude that the patient suffering from type II disease (denoted GM1260) carried both mutations described above. Any one of the amino acid changes described reduces the glucocerebrosidase activity as tested by transfection of COS cells with expression vectors harboring the mutated cDNAs. The base changes in the two mutated cDNAs do not affect the electrophoretic mobility of the corresponding polypeptides on an SDS polyacrylamide gel.
-
(1989) Genomics. 4, 1, p. 87-96 Abstract
We report the sequence of the entire human gene encoding β-glucocerebrosidase and that of the associated pseudogene. The gene contains 11 exons extending from base pair 355 to base pair 7232 in the overall sequence. The gene promoter contains TATA-and CAT-like boxes upstream of the major 5 end of the glucocerebrosidase RNA. The two TATA boxes lie between nucleotides (-23)-(-27) and (-33)-(-39) and the two possible CAT boxes reside between nucleotides (-90)-(-94) and (-96)-(-99) in relation to the major 5 end of the mRNA. The functionality of the promoter region was monitored by coupling it to the bacterial gene coding for chloramphenicol acetyltransferase (CAT) and assaying the expression of the enzyme in cells transfected with this vector. The glucocerebrosidase promoter not only directs synthesis of the bacterial enzyme but also exhibits the same pattern of tissue-specific expression as that of the endogenous gene. An apparently tightly linked pseudogene is approximately 96% homologous to the functional gene. However, introns 2, 4, 6, and 7 have large "deletions" consisting of Alu sequences 313, 626, 320, and 277 bp in length, respectively. It is entirely possible that the ancestral gene lacks these sequences and that they have been inserted into the introns of the functioning gene. There is also a 55-bp deletion from a part of exon 9 flanked by a short inverted repeat. The sequence data should facilitate development of methods for diagnosis of Gaucher disease at the molecular level.
1988
-
(1988) Gene. 73, 2, p. 469-478 Abstract
Gaucher disease is an inborn error of sphingolipid metabolism. It is due to decreased enzymatic activity of glucocerebrosidase (GCase) which causes accumulation of glucocerebrosides, mainly in cells of the reticulo-endothelial system. The disorder is clinically heterogenous and can include central nervous system signs. However, the manifestations of the disease in most cases are restricted to a limited number of cell types and organs. This could be explained by highly differential expression of the human gcs gene. To test this notion, the level of GCase-specific mRNA was determined m different human cell lines by hybridizing Northern blots to a human GCase-specific cDNA probe or by using the RNase protection method. It was found that epithelial cells exhibit high levels of GCase mRNA while skin fibroblasts and promyelocytes show intermediate steadystate levels of this RNA. Macrophages have low steady-state levels of GCase mRNA and in B-cells it is hardly detectable. Moreover, when B-cells or skin fibroblasts were transfected with a vector harbouring the bacterial cat gene coupled to the human gcs gene promoter, the levels of CAT expressed in each cell type were directly correlated to the amount of endogenous GCase RNA. Comparison of the GCase mRNA levels in Gaucher-versus non-Gaucher-derived cells revealed that in Gaucher cells this RNA is always more abundant than in the corresponding non-Gaucher counterparts, suggesting the involvement of a feed-back mechanism sensitive to the levels of actual enzymatic activity.
-
(1988) DNA. 7, 2, p. 107-116 Abstract
Two different genomic clones containing the entire coding sequence of human glucocerebrosidase were isolated from a fetal liver library using a cDNA probe previously cloned by us. These clones correspond to two human glucocerebrosidase genes, designated 6-1 and 10-2. Clone 6-1 contains sequences homologous to the cDNA we cloned previously. The promoter regions of the genes were identified by S1 analysis and sequenced. They contain TATA- and CAAT-like boxes, but lack a GGCGGG motif. When coupled to the bacterial gene coding for chloramphenicol acetyl transferase (CAT) and transfected to Gaucher skin fibroblast lines, both promoter fragments enhanced CAT activity. The promoter of gene 6-1 was eight times more efficient than the promoter of gene 10-2. Northern blot analysis revealed three human glucocerebrosidase RNA species of 6, 2.6, and 2.2 kb in size. The 6-kb transcript is probably a nuclear transcript whereas the 2.6-kb and 2.2-kb transcripts are cytoplasmic species which emerge from polyadenylation at different sites.
1987
-
(1987) DNA. 6, 2, p. 101-108 Abstract
A human glucocerebrosidase cDNA clone was isolated from a human chronic myelogenous leukemia (line K562) cDNA library using a 36-nucleotide-long synthetic probe (GC-36). The 2.4-kb cDNA contains 184 bp of 5 nontranslated sequences, the complete coding region, and 546 bp of 3 nontranslated sequences followed by 100 bp of poly(A). A primer extension experiment indicated that the cDNA is at least 51 bp shorter than the mRNA at the 5 end. In normal human placenta as well as in fibroblasts from Gaucher's disease patients, a major mRNA species of 2.6 kb hybridizes with the cDNA probe. The amounts of the glucocerebrosidase mRNA in normal placenta and Gaucher's cells are comparable. The cDNA was linked to the SP6 promoter and transcribed in vitro. The resultant RNA, when translated in a cell-free system, yielded a polypeptide of 55 kD, which is the size expected from the coding sequence. The cDNA was inserted into an SV40 shuttle vector, under the transcription control of the SV40 early promoter. COS-M6 cells were transfected with this construct and the biological activity of the cDNA was assayed by monitoring the increase in glucocerebrosidase activity, using 4-methyl umbiliferyl glucopyranoside as a substrate. There was a two- to three-fold increase in enzymatic activity in the transfected cells, compared to nontransfected cells. These results prove the authenticity of the glucocerebrosidase cDNA and provide the basis for experiments to understand the nature of the genetic alterations responsible for Gaucher's disease.