We love to understand and explain the world using numbers as our sixth sense. Our group brings the tools of systems biology to bear on the grand challenges of sustainability. My lab members and I are passionate about trying to understand the cellular highways of energy and carbon transformations known as central carbon metabolism in quantitative terms. We employ a combination of computational and experimental synthetic biology tools.
Rosenberg Y., Wiedenhofer D., Virág D., Bar-Sella G., Greenspoon L., Herrnstadt B., Akenji L., Phillips R. & Milo R.
(2025)
Nature Ecology and Evolution.
9,
12,
p. 2259-2264
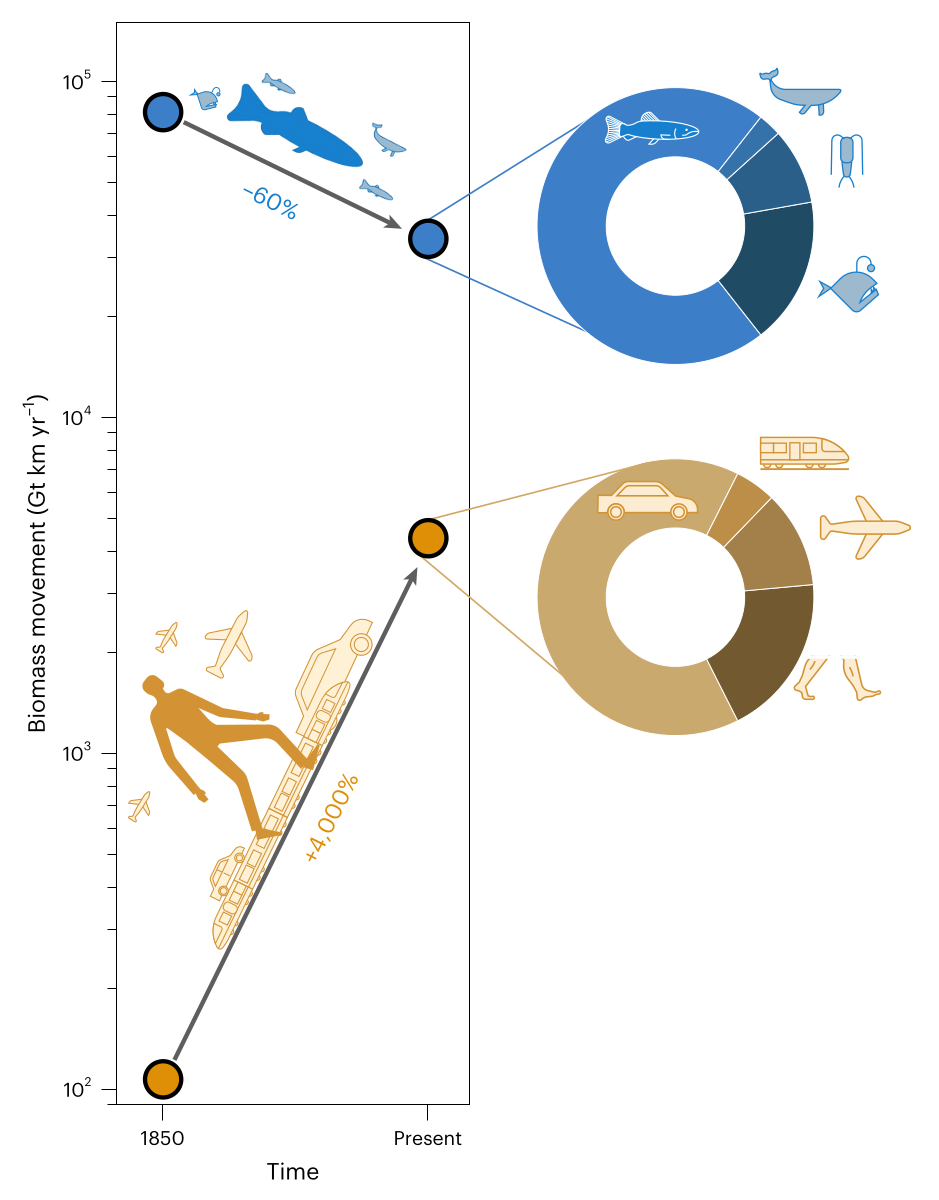
Earth is teeming with life on the move, shaping ecosystems and human civilizations alike. However, the magnitude of movement by humans and other animals has yet to be assessed holistically. Here we quantify the movement of biomass across all animal life and in comparison to humanity. We show that the combined biomass movement of all wild birds, land arthropods and wild land mammals is about one-sixth that of humans walking and about 40 times smaller than all the biomass movement of humans. The biomass movement of marine animals, which we find to be the living worlds largest, has been halved since 1850 due to industrial fishing and whaling, while human biomass movement has increased by about 40-fold. This study gives a quantitative perspective on global mobility in the Anthropocene and sharpens our perception regarding the extent of human versus animal activity.
Mammals are of central interest in ecology and conservation science. Here, we estimate the trajectory of mammal biomass globally over time including humans, domesticated and wild mammals. According to our estimates, in the 1850s, the combined biomass of wild mammals was ≈200 Mt (million tonnes), roughly equal to that of humanity and its domesticated mammals at that time. Since then, human and domesticated mammal populations have grown rapidly, reaching their current combined biomass of ≈1100 Mt. During the same period, the total biomass of wild mammals decreased by more than 2-fold. We estimate that, despite a moderate increase in the recent decades, the global biomass of wild marine mammals has declined by ≈70% since the 1850s. This provides a broader perspective to observed species extinctions, with ≈2% of marine mammal species recorded as extinct during the same period. While historical wild mammal biomass estimates rely on limited data and have various uncertainties, they provide a complementary perspective to species extinctions and other metrics in tracking the status of wildlife. This work additionally provides a quantitative view on the rapid human-induced shift in the composition of mammalian biomass over the past two centuries.
de Pins B., Malbranke C., Jabłońska J., Shmuel A., Sharon I., Bitbol A. F., Mueller-Cajar O., Noor E. & Milo R.
(2025)
Proceedings of the National Academy of Sciences - PNAS.
122,
47,
e250143312.
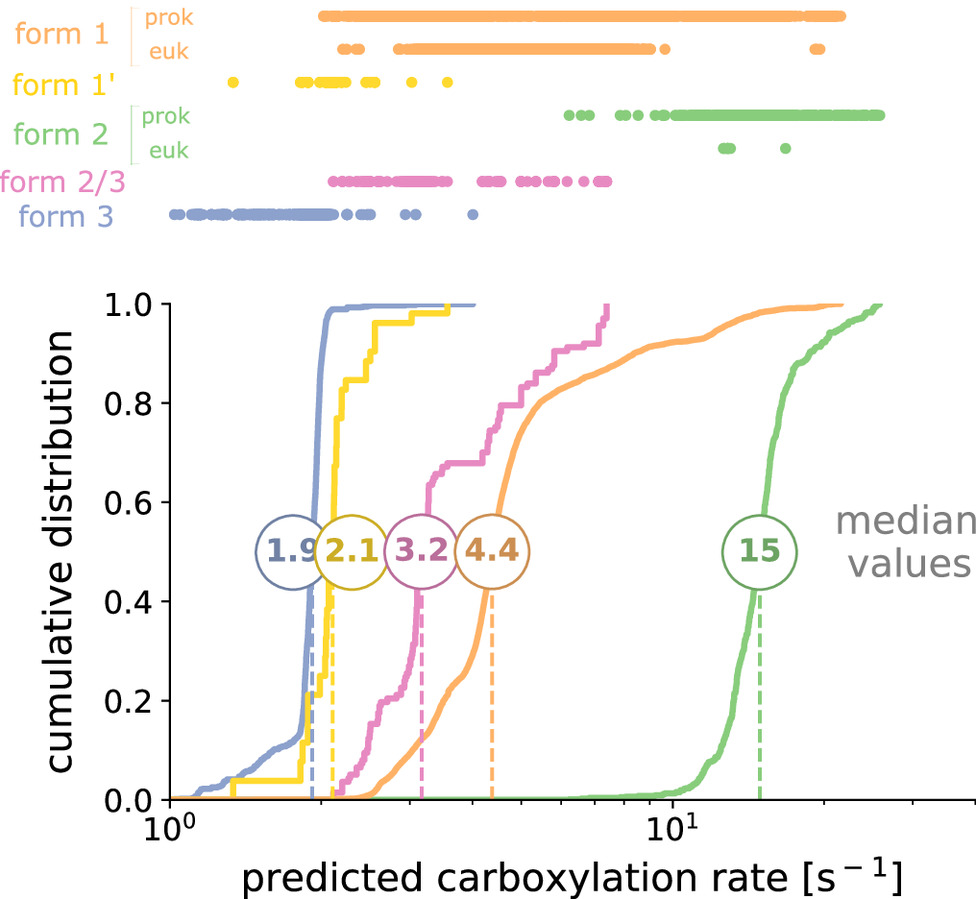
Rubisco is the main gateway through which inorganic carbon enters the biosphere, catalyzing the vast majority of carbon fixation on Earth. This pivotal enzyme has long been observed to be kinetically constrained. Yet, this impression is based on kinetic measurements heavily focused on eukaryotic rubiscos, a rather conserved group of low genetic diversity. Moreover, the fastest rubiscos that we know of so far were found among the sparsely sampled prokaryotes. Could there be yet faster rubiscos among the uncharted regions of rubiscos phylogenetic diversity? Here, we perform a characterization of more than 250 rubiscos from a wide range of bacteria and archaea, thereby doubling the coverage of the diversity of this key enzyme. We assess the distribution of the carboxylation rates at saturating levels of CO2, and establish that rubisco is a relatively slow enzyme across the tree of life, never exceeding ≈30 reactions per second at 30 °C. We show that relatively faster subclades share similar evolutionary contexts, involving micro-oxygenic environments or a CO2 concentrating mechanism. Leveraging a simple machine learning model trained on this dataset, we predict the carboxylation rate for all ≈68,000 sequenced rubisco variants found in nature to date. This study provides the largest and most diverse dataset of natural variants for an enzyme and their associated rates, establishing a solid benchmark for future efforts to predict catalytic rates from sequence data.
Lovat S. J., Ben-Nissan R., Milshtein E., Tzachor A., Flamholz A., Leger D., Noor E. & Milo R.
(2025)
Nature Biotechnology.
43,
6,
p. 848-853
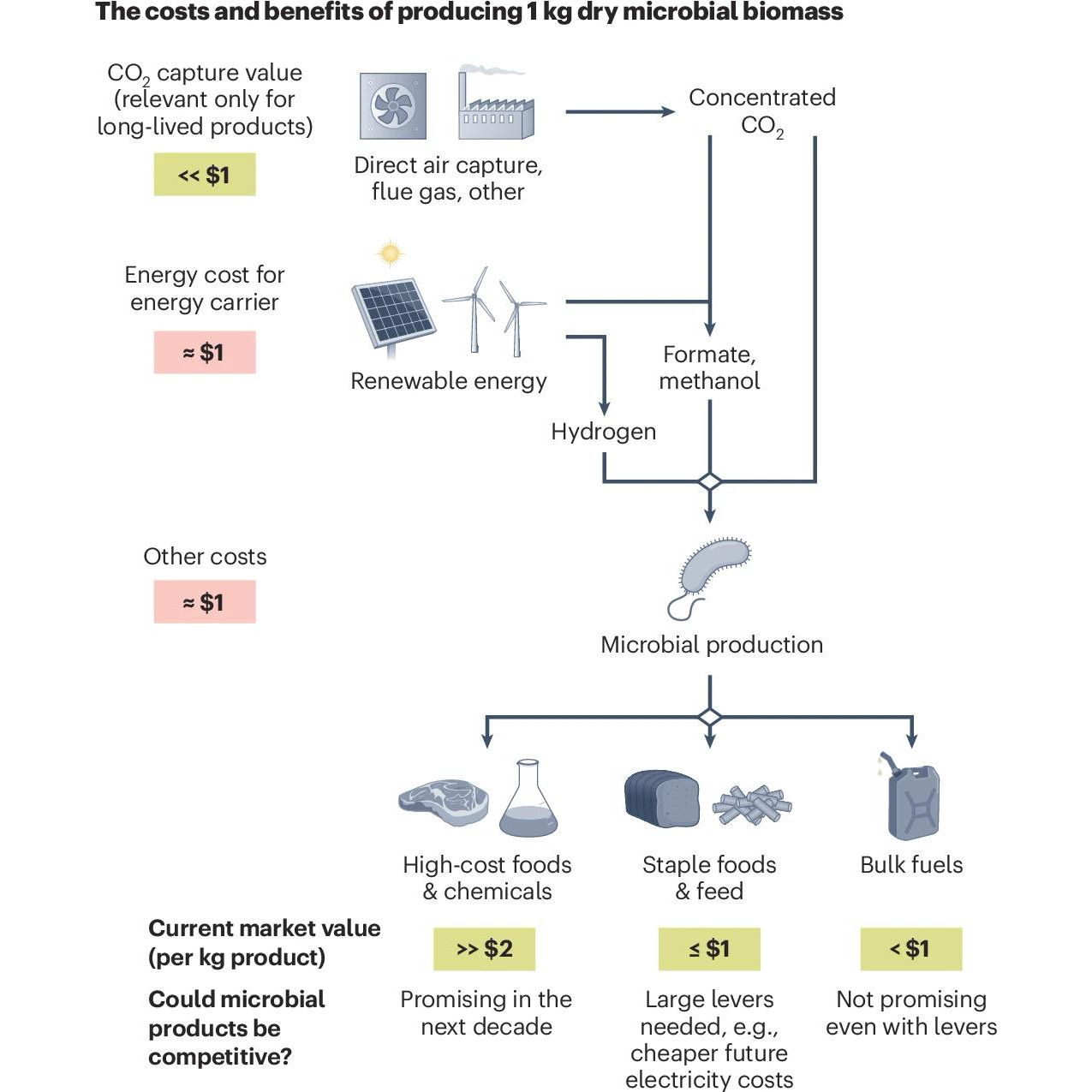
Producing goods, such as foods and fuels, with minimal environmental impacts is urgently needed. Although advances in bioproduction are promising, there is often a noticeable gap in our understanding of whether and where new processes can compete with existing methods on an economic and environmental basis. Transparent lower bound calculations from basic principles highlight potential benefits of producing foods, but not fuels, from electro-microbial production of biomass.
de Pins B., Greenspoon L., Bar-On Y. M., Shamshoum M., Ben-Nissan R., Milshtein E., Davidi D., Sharon I., Mueller-Cajar O., Noor E. & Milo R.
(2024)
EMBO Journal.
43,
14,
p. 3072-3083
Autotrophy is the basis for complex life on Earth. Central to this process is rubiscothe enzyme that catalyzes almost all carbon fixation on the planet. Yet, with only a small fraction of rubisco diversity kinetically characterized so far, the underlying biological factors driving the evolution of fast rubiscos in nature remain unclear. We conducted a high-throughput kinetic characterization of over 100 bacterial form I rubiscos, the most ubiquitous group of rubisco sequences in nature, to uncover the determinants of rubiscos carboxylation velocity. We show that the presence of a carboxysome CO2 concentrating mechanism correlates with faster rubiscos with a median fivefold higher rate. In contrast to prior studies, we find that rubiscos originating from α-cyanobacteria exhibit the highest carboxylation rates among form I enzymes (≈10 s−1 median versus
Ben Nissan R., Milshtein E., Pahl V., de Pins B., Jona G., Levi D., Yung H., Nir N., Ezra D., Gleizer S., Link H., Noor E. & Milo R.
(2024)
eLife.
12,
RP88793.
Synthetic autotrophy is a promising avenue to sustainable bioproduction from CO2. Here, we use iterative laboratory evolution to generate several distinct autotrophic strains. Utilising this genetic diversity, we identify that just three mutations are sufficient for Escherichia coli to grow autotrophically, when introduced alongside non-native energy (formate dehydrogenase) and carbon-fixing (RuBisCO, phosphoribulokinase, carbonic anhydrase) modules. The mutated genes are involved in glycolysis (pgi), central-carbon regulation (crp), and RNA transcription (rpoB). The pgi mutation reduces the enzyme's activity, thereby stabilising the carbon-fixing cycle by capping a major branching flux. For the other two mutations, we observe down-regulation of several metabolic pathways and increased expression of native genes associated with the carbon-fixing module (rpiB) and the energy module (fdoGH), as well as an increased ratio of NADH/NAD+ - the cycle's electron-donor. This study demonstrates the malleability of metabolism and its capacity to switch trophic modes using only a small number of genetic changes and could facilitate transforming other heterotrophic organisms into autotrophs.
Greenspoon L., Krieger E., Sender R., Rosenberg Y., Bar-On Y. M., Moran U., Antman T., Meiri S., Roll U., Noor E. & Milo R.
(2023)
Proceedings of the National Academy of Sciences of the United States of America.
120,
10,
e220489212.
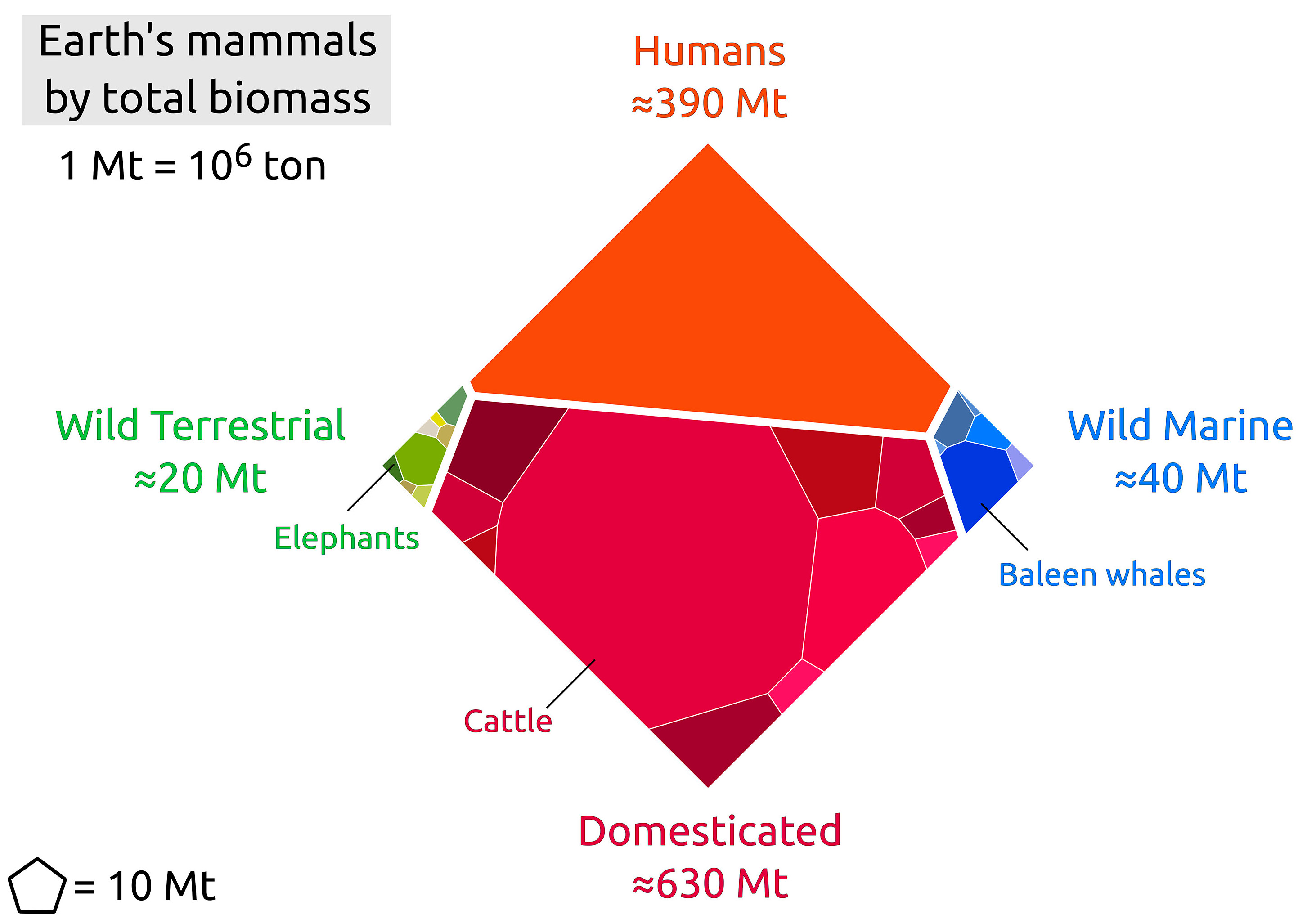
Wild mammals are icons of conservation efforts, yet there is no rigorous estimate available for their overall global biomass. Biomass as a metric allows us to compare species with very different body sizes, and can serve as an indicator of wild mammal presence, trends, and impacts, on a global scale. Here, we compiled estimates of the total abundance (i.e., the number of individuals) of several hundred mammal species from the available data, and used these to build a model that infers the total biomass of terrestrial mammal species for which the global abundance is unknown. We present a detailed assessment, arriving at a total wet biomass of ≈20 million tonnes (Mt) for all terrestrial wild mammals (95% CI 13-38 Mt), i.e., ≈3 kg per person on earth. The primary contributors to the biomass of wild land mammals are large herbivores such as the white-tailed deer, wild boar, and African elephant. We find that even-hoofed mammals (artiodactyls, such as deer and boars) represent about half of the combined mass of terrestrial wild mammals. In addition, we estimated the total biomass of wild marine mammals at ≈40 Mt (95% CI 20-80 Mt), with baleen whales comprising more than half of this mass. In order to put wild mammal biomass into perspective, we additionally estimate the biomass of the remaining members of the class Mammalia. The total mammal biomass is overwhelmingly dominated by livestock (≈630 Mt) and humans (≈390 Mt). This work is a provisional census of wild mammal biomass on Earth and can serve as a benchmark for human impacts.