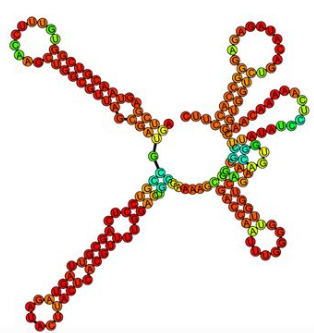
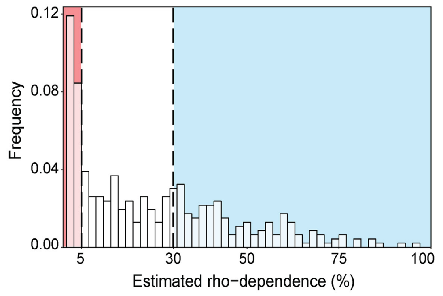
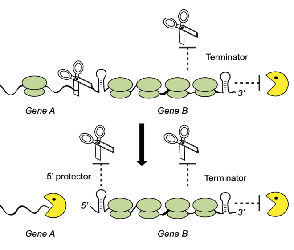
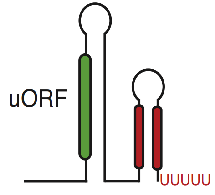
Millman A., Melamed S., Leavitt A., Doron S., Bernheim A., Hör J., Garb J., Bechon N., Brandis A., Lopatina A., Ofir G., Hochhauser D., Stokar-Avihail A., Tal N., Sharir S., Voichek M., Erez Z., Ferrer J. L. M., Dar D., Kacen A., Amitai G. & Sorek R.
(2022)
Cell Host and Microbe.
30,
11,
p. 1556-1569.e5
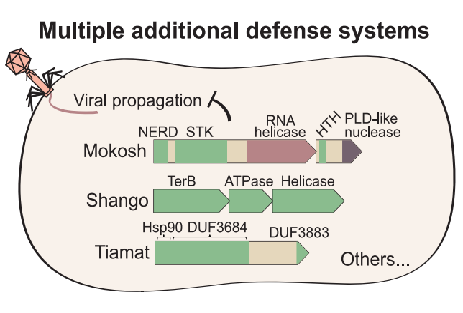
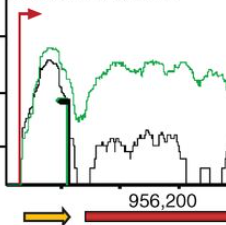
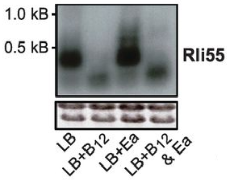
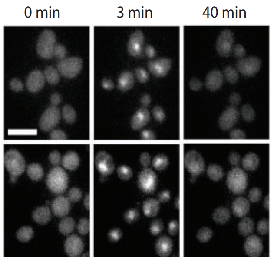
Bacterial anti-phage systems are frequently clustered in microbial genomes, forming defense islands. This property enabled the recent discovery of multiple defense systems based on their genomic co-localization with known systems, but the full arsenal of anti-phage mechanisms remains unknown. We report the discovery of 21 defense systems that protect bacteria from phages, based on computational genomic analyses and phage-infection experiments. We identified multiple systems with domains involved in eukaryotic antiviral immunity, including those homologous to the ubiquitin-like ISG15 protein, dynamin-like domains, and SEFIR domains, and show their participation in bacterial defenses. Additional systems include domains predicted to manipulate DNA and RNA molecules, alongside toxin-antitoxin systems shown here to function in anti-phage defense. These systems are widely distributed in microbial genomes, and in some bacteria, they form a considerable fraction of the immune arsenal. Our data substantially expand the inventory of defense systems utilized by bacteria to counteract phage infection.