Sphingolipids play a crucial role in cell membrane function. Using experimental and computational techniques, the Futerman laboratory studies the mechanisms that drive the generation of sphingolipid diversity, the roles of sphingolipid complexity in membrane structure and function, and evolutionary processes that may, or may not, explain the emergence of such complexity.
Futerman A. H.
(2024)
Pflugers Archiv European Journal of Physiology.
476,
12,
p. 1781-1788
Research on sphingolipids has proliferated exponentially over the past couple of decades, as exemplified in the findings reported at the International Leopoldina Symposium on Lipid Signaling held in Frankfurt in late 2023. Most researchers in the field study how sphingolipids function in regulating a variety of cellular processes and, in particular, how they are dysregulated in numerous human diseases; however, I now propose that we implement a more holistic research program in our study of sphingolipids, which embraces a sense of awe and wonder at the complexities and beauty of sphingolipids and of sphingolipid metabolism. I will outline the chemical complexity of sphingolipids, their modes of interaction within the lipid bilayer, and their biosynthetic pathways. I will then briefly touch upon the ability of current neo-Darwinian mechanisms to explain the emergence of both sphingolipids and of the complex pathways that generate them. Although such discussion is normally considered taboo in biological circles, I nevertheless submit that in-depth analysis of the minutiae of metabolic pathways, such as those of the sphingolipid biosynthetic pathway, raises challenges to current neo-Darwinian mechanisms that should not be shunned or ignored.
Milenkovic I., Blumenreich S., Hochfelder A., Azulay A., Biton I. E., Zerbib M., Oren R., Tsoory M., Joseph T., Fleishman S. J. & Futerman A. H.
(2024)
Gene Therapy.
31,
9-10,
p. 439-444
Almost all attempts to date at gene therapy approaches for monogenetic disease have used the amino acid sequences of the natural protein. In the current study, we use a designed, thermostable form of glucocerebrosidase (GCase), the enzyme defective in Gaucher disease (GD), to attempt to alleviate neurological symptoms in a GD mouse that models type 3 disease, i.e. the chronic neuronopathic juvenile subtype. Upon injection of an AAVrh10 (adeno-associated virus, serotype rh10) vector containing the designed GCase (dGCase) into the left lateral ventricle of Gba−/−;Gbatg mice, a significant improvement in body weight and life-span was observed, compared to injection of the same mouse with the wild type enzyme (wtGCase). Moreover, a reduction in levels of glucosylceramide (GlcCer), and an increase in levels of GCase activity were seen in the right hemisphere of Gba−/−;Gbatg mice, concomitantly with a significant improvement in motor function, reduction of neuroinflammation and a reduction in mRNA levels of various genes shown previously to be elevated in the brain of mouse models of neurological forms of GD. Together, these data pave the way for the possible use of modified proteins in gene therapy for lysosomal storage diseases and other monogenetic disorders.
Biran A., Santos T. C., Dingjan T. & Futerman A. H.
(2024)
Biochimica et Biophysica Acta - Molecular and Cell Biology of Lipids.
1869,
3,
159462.
In eukaryotes, the de novo synthesis of sphingolipids (SLs) consists of multiple sequential steps which are compartmentalized between the endoplasmic reticulum and the Golgi apparatus. Studies over many decades have identified the enzymes in the pathway, their localization, topology and an array of regulatory mechanisms. However, little is known about the evolutionary forces that underly the generation of this complex pathway or of its anteome, i.e., the metabolic pathways that converge on the SL biosynthetic pathway and are essential for its activity. After briefly describing the pathway, we discuss the mechanisms by which the enzymes of the SL biosynthetic pathway are targeted to their different subcellular locations, how the pathway per se may have evolved, including its compartmentalization, and the relationship of the pathway to eukaryogenesis. We discuss the circular interdependence of the evolution of the SL pathway, and comment on whether current Darwinian evolutionary models are able to provide genuine mechanistic insight into how the pathway came into being.
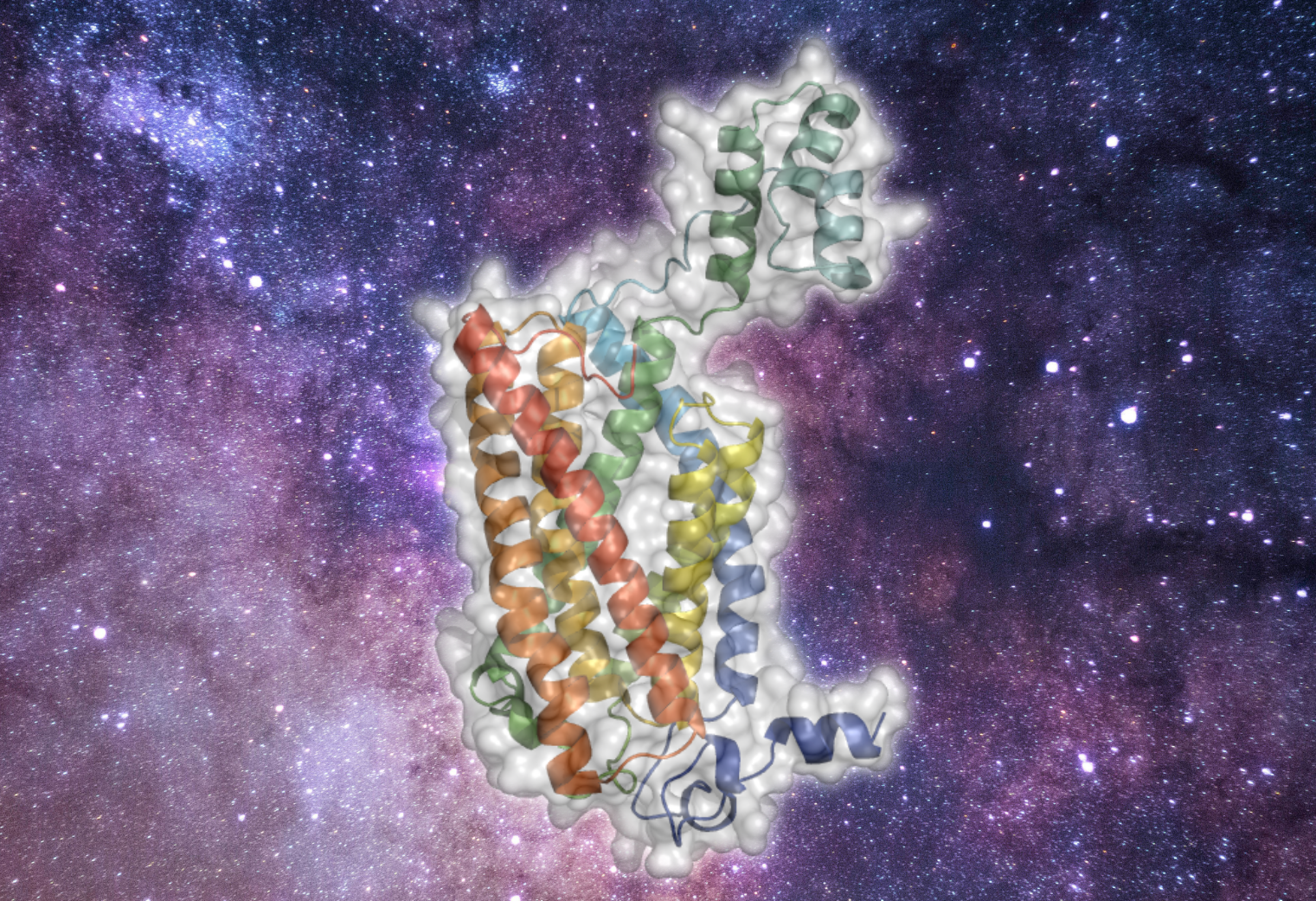
Zelnik I. D., Mestre B., Weinstein J. J., Dingjan T., Izrailov S., Ben-Dor S., Fleishman S. J. & Futerman A. H.
(2023)
Nature Communications.
14,
1,
2330.
Until now, membrane-protein stabilization has relied on iterations of mutations and screening. We now validate a one-step algorithm, mPROSS, for stabilizing membrane proteins directly from an AlphaFold2 model structure. Applied to the lipid-generating enzyme, ceramide synthase, 37 designed mutations lead to a more stable form of human CerS2. Together with molecular dynamics simulations, we propose a pathway by which substrates might be delivered to the ceramide synthases.
Dingjan T. & Futerman A. H.
(2021)
BioEssays.
43,
5,
e2100021.
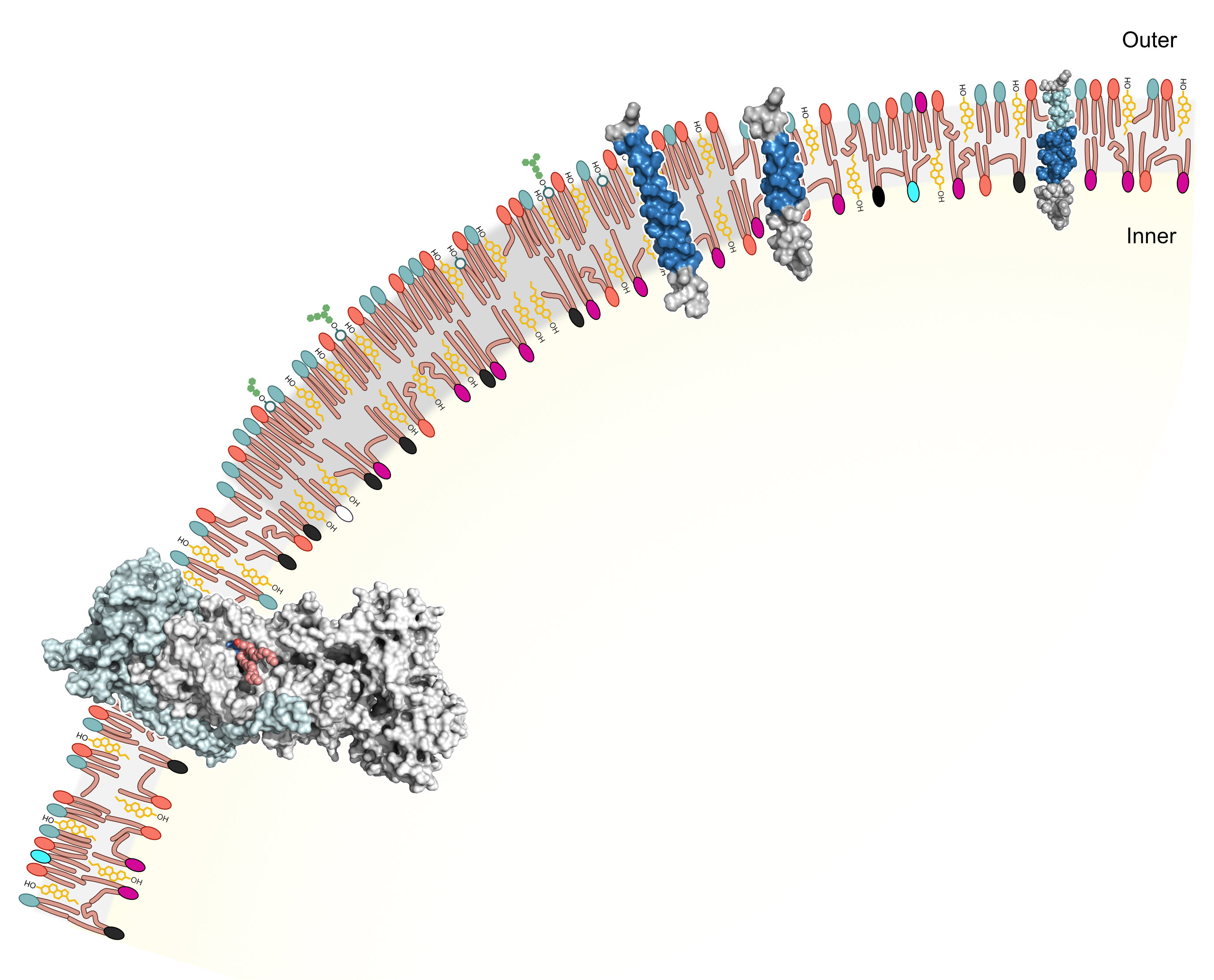
Cell membranes are now emerging as finely tuned molecular systems, signifying that reevaluation of our understanding of their structure is essential. Although the idea that cell membrane lipid bilayers do little more than give shape and form to cells and limit diffusion between cells and their environment is totally passé, the structural, compositional, and functional complexity of lipid bilayers often catches cell and molecular biologists by surprise. Models of lipid bilayer structure have developed considerably since the heyday of the fluid mosaic model, principally by the discovery of the restricted diffusion of membrane proteins and lipids within the plane of the bilayer. In reviewing this field, we now suggest that further refinement of current models is necessary and propose that describing lipid bilayers as \u201cfinelytuned molecular assemblies\u201d best portrays their complexity and function.