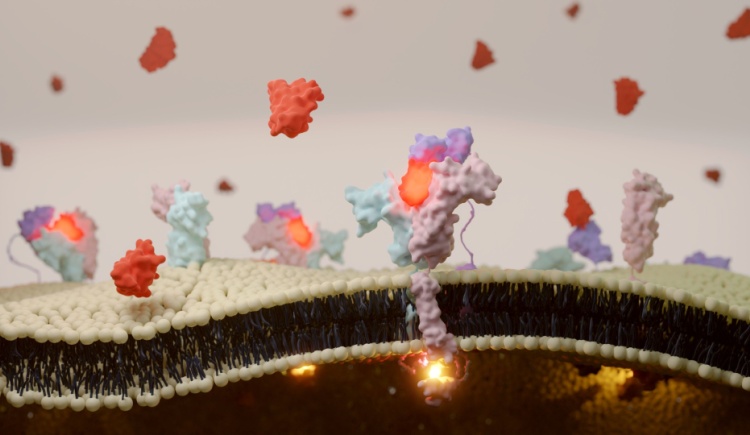
Our project tackles a fundamental obstacle impeding the clinical advancement of RNA therapies: their delivery. We are investigating extracellular vesicles (EVs) as an ideal carrier for biological macromolecules, such as coding RNA, due to their natural biocompatibility and potential for customization, given their origin from human cells. Our objective is to develop a flexible system for the efficient encapsulation of mRNA within EVs. We have already established a pioneering technique that allows cells to generate EVs tailored to engage with particular receptors on target cells. Advancing from this groundwork, we are now introducing innovative methods to actively load mRNA into EVs and direct them toward specific organs, with a focus on the lungs. This initiative promises to not only deepen our insight into RNA localization mechanisms but also to forge a targeted, EV-centric delivery method for RNA therapeutics, representing a considerable leap forward in the field of personalized medicine