Rotem A., Kaplan Y., Gefen O., Ronin I., Gutfreund A., Rappeport H., Faigenbaum-Romm R., Naor N., Stav E., Agam O. & Balaban N. Q.
(2026)
Science advances.
12,
1,
p. eadt6577
eadt6577.
Antibiotic persistence, typically attributed to dormant bacteria, is known to be a major cause of treatment failure. However, despite many years of intense research, no clear consensus on its mechanism has emerged. Here, we demonstrate that high survival under antibiotics may originate from two fundamentally different growth-arrest archetypes: either from a regulated growth arrest, leading to a protected dormant cellular state, or from a dysregulated disrupted growth arrest. Using modeling and experimental approaches including transcriptomics, microcalorimetry, and microfluidics, we unveil the characteristics and vulnerabilities of each growth-arrest archetype. In particular, disrupted bacteria show a general impairment of membrane homeostasis. This understanding resolves previous conflicting results regarding characteristics of persisters and allows tailoring treatments that target the different growth-arrested bacteria. The fundamental distinction between regulated and disrupted growth arrests should be broadly relevant for the description of cells under stress.
Aroeti L., Elbaz N., Faigenbaum-Romm R., Yakovian O., Altuvia Y., Argaman L., Katsowich N., Bejerano-Sagie M., Ravins M., Margalit H., Ben-Yehuda S. & Rosenshine I.
(2025)
Nature Communications.
16,
1,
3834.
The RNA-binding protein CsrA regulates the expression of hundreds of genes in several bacterial species, thus controlling virulence and other processes. However, the outcome of the CsrA-mRNA interactions is modulated by competing small RNAs and other factors through mechanisms that are only partially understood. Here, we show that CsrA accumulates in a dynamic membraneless compartment in cells of E. coli and other pathogenic species. In addition to CsrA, the compartment contains components of the RNA-degrading complex (degradosome), regulatory small RNAs, and selected mRNAs. Formation of the compartment is associated with a switch between promoting and repressing virulence gene expression by CsrA. We suggest that similar CsrA switches may be widespread in diverse bacteria.
Faigenbaum-Romm R., Yedidi N., Gefen O., Katsowich-Nagar N., Aroeti L., Ronin I., Bar-Meir M., Rosenshine I. & Balaban N. Q.
(2025)
Cell.
188,
19,
p. 5313-5331.e18
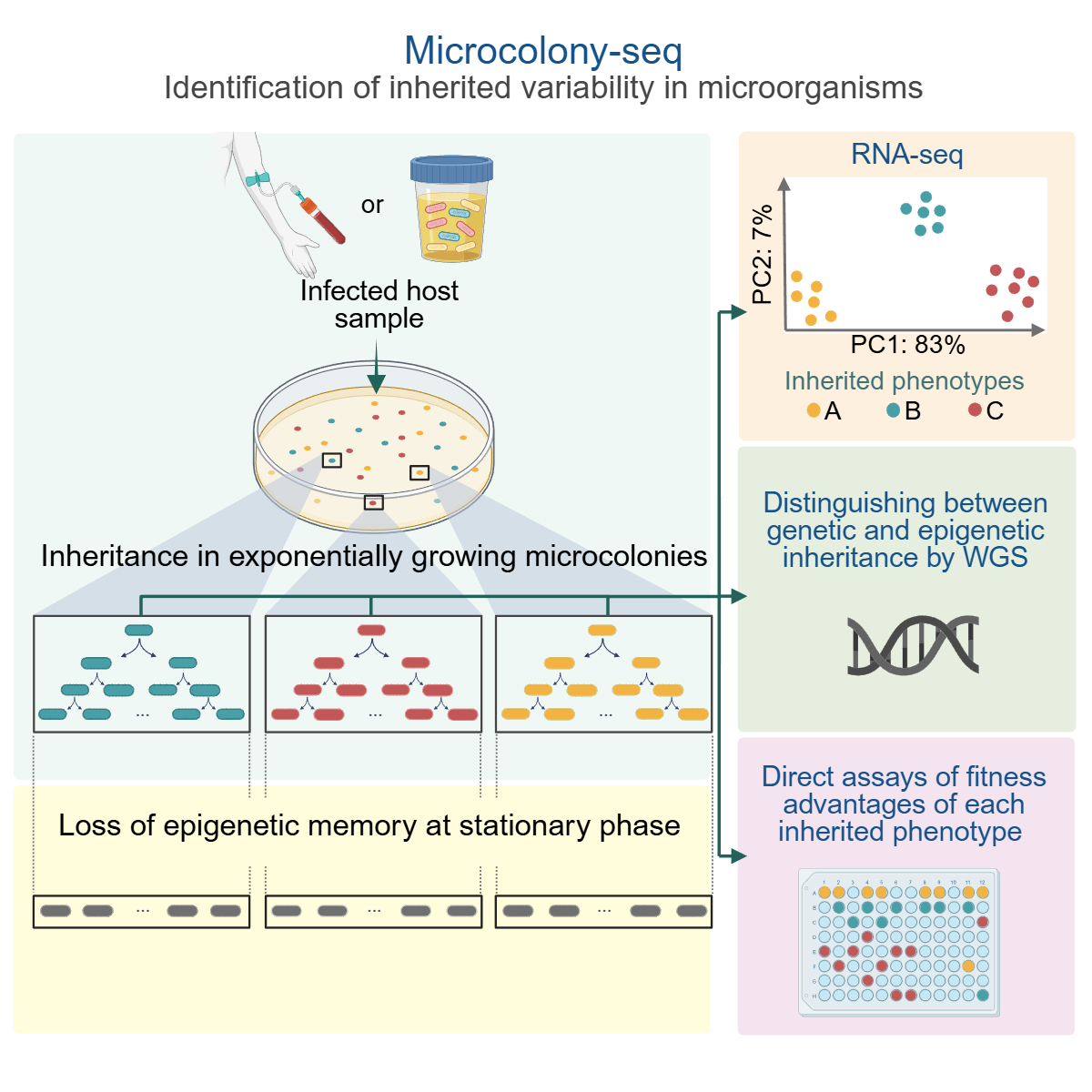
Uncovering phenotypic heterogeneity is fundamental to understanding processes such as development and stress responses. Due to the low mRNA abundance in single bacteria, determining biologically relevant heterogeneity remains a challenge. Using Microcolony-seq, a methodology that captures inherited heterogeneity by analyzing microcolonies originating from single bacterial cells, we uncover the ubiquitous ability of bacteria to maintain long-term inheritance of the host environment. Notably, we observe that growth to stationary phase erases the epigenetic inheritance. By leveraging this memory within each microcolony, Microcolony-seq combines bulk RNA sequencing (RNA-seq) with whole-genome sequencing and phenotypic assays to detect the distinct subpopulations and their fitness advantages. Applying this directly to infected human samples enables us to uncover a wealth of diverse inherited phenotypes. Our observations suggest that bacterial memory may be a widespread phenomenon in both Gram-negative and Gram-positive bacteria. Microcolony-seq provides potential targets for the rational design of therapies with the power to simultaneously target the coexisting subpopulations.
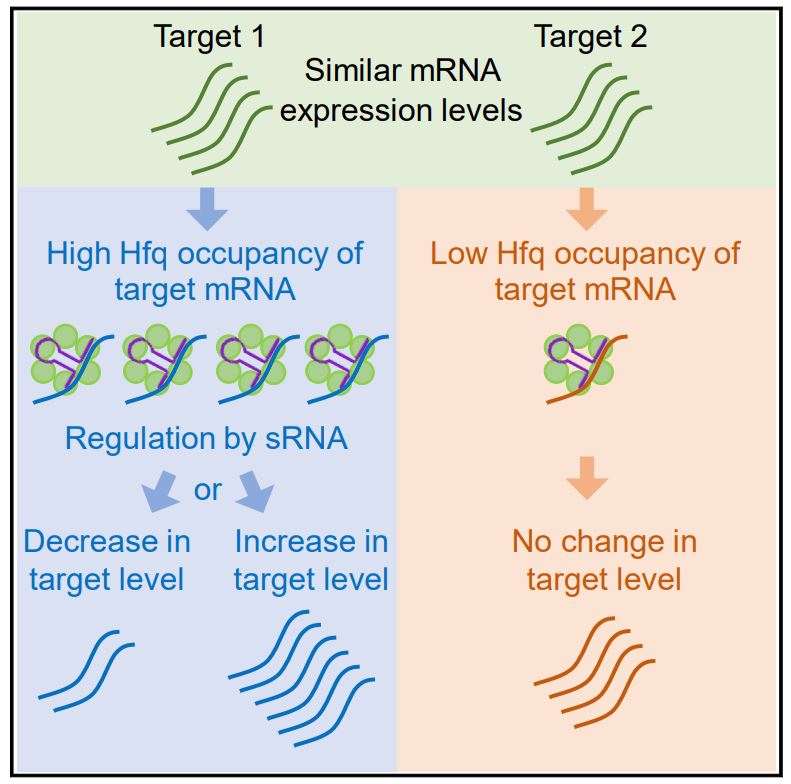
Faigenbaum-Romm et al. analyze data of sRNA-target pairs and transcriptome measurements, revealing that only a subset of targets shows expression changes under overexpression of the sRNA. Analyzing various target features, they find that competition among targets over binding the chaperon Hfq plays a major role in the regulatory outcome.
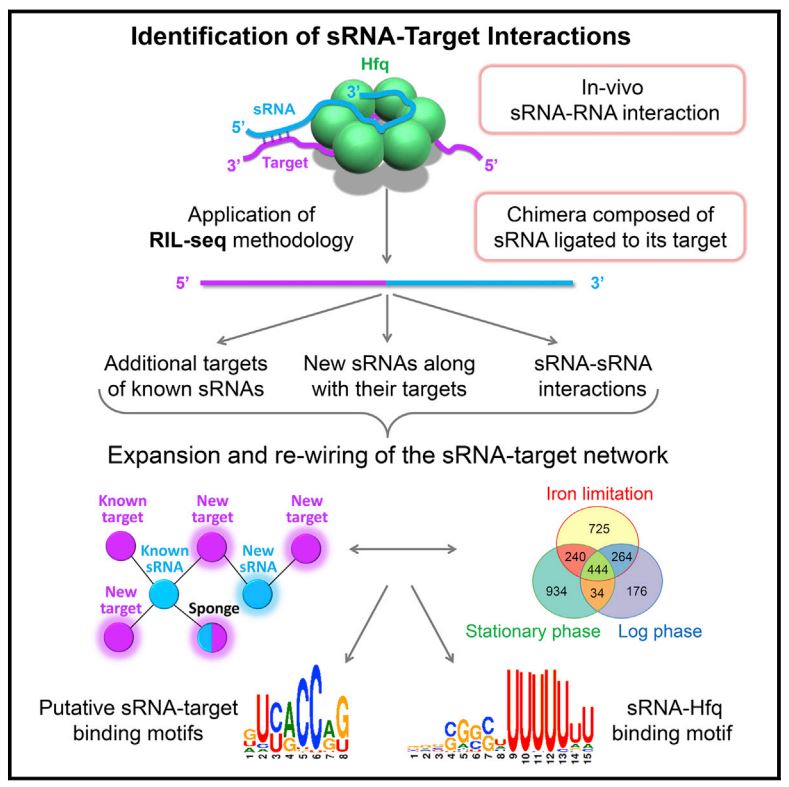
Small RNAs (sRNAs) are major post-transcriptional regulators of gene expression in bacteria. To enable transcriptome-wide mapping of bacterial sRNA-target pairs, we developed RIL-seq (RNA interaction by ligation and sequencing). RIL-seq is an experimental-computational methodology for capturing sRNA-target interactions in vivo that takes advantage of the mutual binding of the sRNA and target RNA molecules to the RNA chaperone protein Hfq. The experimental part of the protocol involves co-immunoprecipitation of Hfq and bound RNAs, ligation of RNAs, library preparation and sequencing. The computational pipeline maps the sequenced fragments to the genome, reveals chimeric fragments (fragments comprising two ligated independent fragments) and determines statistically significant overrepresented chimeric fragments as interacting RNAs. The statistical filter is aimed at reducing the number of spurious interactions resulting from ligation of random neighboring RNA fragments, thus increasing the reliability of the determined sRNA-target pairs. A major advantage of RIL-seq is that it does not require overexpression of sRNAs; instead, it simultaneously captures the in vivo targets of all sRNAs in the native state of the cell. Application of RIL-seq to bacteria grown under different conditions provides distinctive snapshots of the sRNA interactome and sheds light on the dynamics and rewiring of the post-transcriptional regulatory network. As RIL-seq needs no prior information about the sRNA and target sequences, it can identify novel sRNAs, along with their targets. It can be adapted to detect protein-mediated RNA-RNA interactions in any bacterium with a sequenced genome. The experimental part of the RIL-seq protocol takes 7-9 d and the computational analysis takes â 1/42 d.
Small RNAs (sRNAs) associated with the RNA chaperon protein Hfq are key posttranscriptional regulators of gene expression in bacteria. Deciphering the sRNA-target interactome is an essential step toward understanding the roles of sRNAs in the cellular networks. We developed a broadly applicable methodology termed RIL-seq (RNA interaction by ligation and sequencing), which integrates experimental and computational tools for in vivo transcriptome-wide identification of interactions involving Hfq-associated sRNAs. By applying this methodology to Escherichia coli we discovered an extensive network of interactions involving RNA pairs showing sequence complementarity. We expand the ensemble of targets for known sRNAs, uncover additional Hfq-bound sRNAs encoded in various genomic regions along with their trans encoded targets, and provide insights into binding and possible cycling of RNAs on Hfq. Comparison of the sRNA interactome under various conditions has revealed changes in the sRNA repertoire as well as substantial re-wiring of the network between conditions.
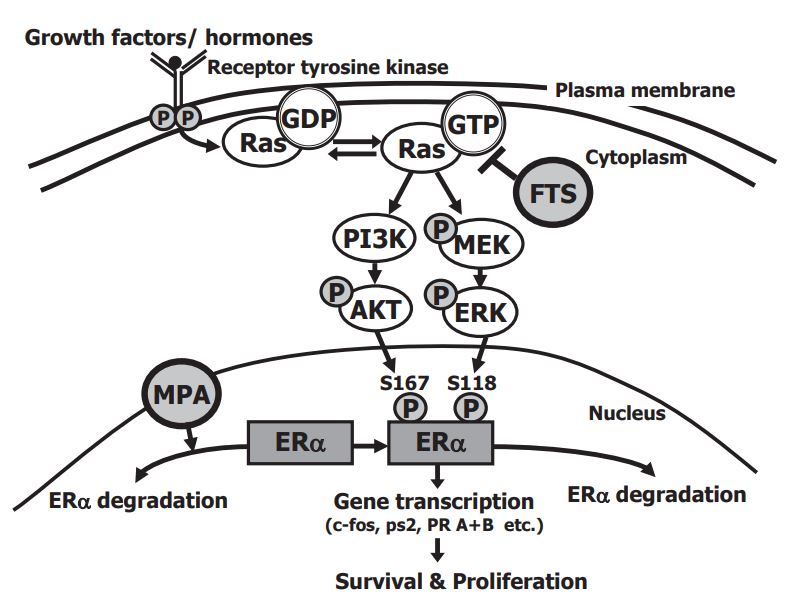
Type 2 endometrial carcinoma (EC) is a poorly differentiated EC. Unlike type 1 EC, which responds to hormonal treatment (progestins), type 2 EC is refractory to hormonal treatment because of its low expression of active estrogen and progesterone receptors (ER, PR). The aim of this study was to develop a novel drug combination designed to treat these aggressive type 2 EC tumors without surgery and with fertility potential preserved. We examined the effects of combined treatment with the progestin medroxyprogesterone acetate (MPA) and the Ras inhibitor S-farnesylthiosalicylic acid (FTS; Salirasib). Because FTS can induce cell differentiation in tumor cells, we examined whether FTS could induce re-differentiation of type 2 EC cells, thereby sensitizing them to MPA. We found that FTS reduced Ras-GTP, phospho- Akt, and phospho-ERK, and that these reductions all correlated with a decrease in ERa phosphorylation. Combined treatment with FTS and MPA induced stronger reduction in USPC1 type 2 EC cell numbers than the reduction induced by either drug alone. MPA caused ERa degradation. Death of the cells was caused by MPA but not by FTS. The phosphorylated ERa induces gene transcription manifested by enhanced cell proliferation and survival. The combination of FTS and MPA, by reducing the mRNA expression of ERa-mediated genes (i.e. PR, c-fos and ps2/TFF1), inhibited tumor growth and enhanced the death of type 2 EC cells. These promising results might herald a novel treatment for the highly aggressive, incurable type 2 endometrial carcinoma.
Elkobi-Peer S., Faigenbaum R. & Carmeli S.
(2012)
Journal of Natural Products.
75,
12,
p. 2144-2151
Five new natural products, aeruginosins GE686 (1), GE766 (2), GE730 (3), GE810 (4), and GE642 (5), were isolated along with four known aeruginosins, 98C, 101, KY642, and DA688, from bloom material of the cyanobacterium Microcystis aeruginosa collected from a fish pond in Kibbutz Geva, Israel, in August 2007. Their structures were elucidated by a combination of various spectroscopic techniques, primarily NMR and MS, while the absolute configurations of the stereogenic centers were determined by Marfey's and chiral-phase HPLC methods. Two of the new aeruginosins, aeruginosins GE686 (1) and GE766 (2), contain the unprecedented d-m-Br-m-Cl-p-hydroxyphenyllactic acid derivative. The structures and biological activities of the five new metabolites are described.