(2016) Science. 351, 6277, p. 1087-1090
Yoav Voichek Lab
Evolution of Transcriptional Regulation

Research
How do plants regulate gene expression to achieve tissue-specific patterns? Does the transcription machinery operate similarly in other eukaryotes, such as fungi and marine algae? What role does the evolution of regulatory sequences play in adaptation to new environments? These and related questions about the evolution of transcriptional regulation lie at the heart of our lab’s interests. To address these, we combine analysis of large genomic datasets with experimental molecular biology.
Research page
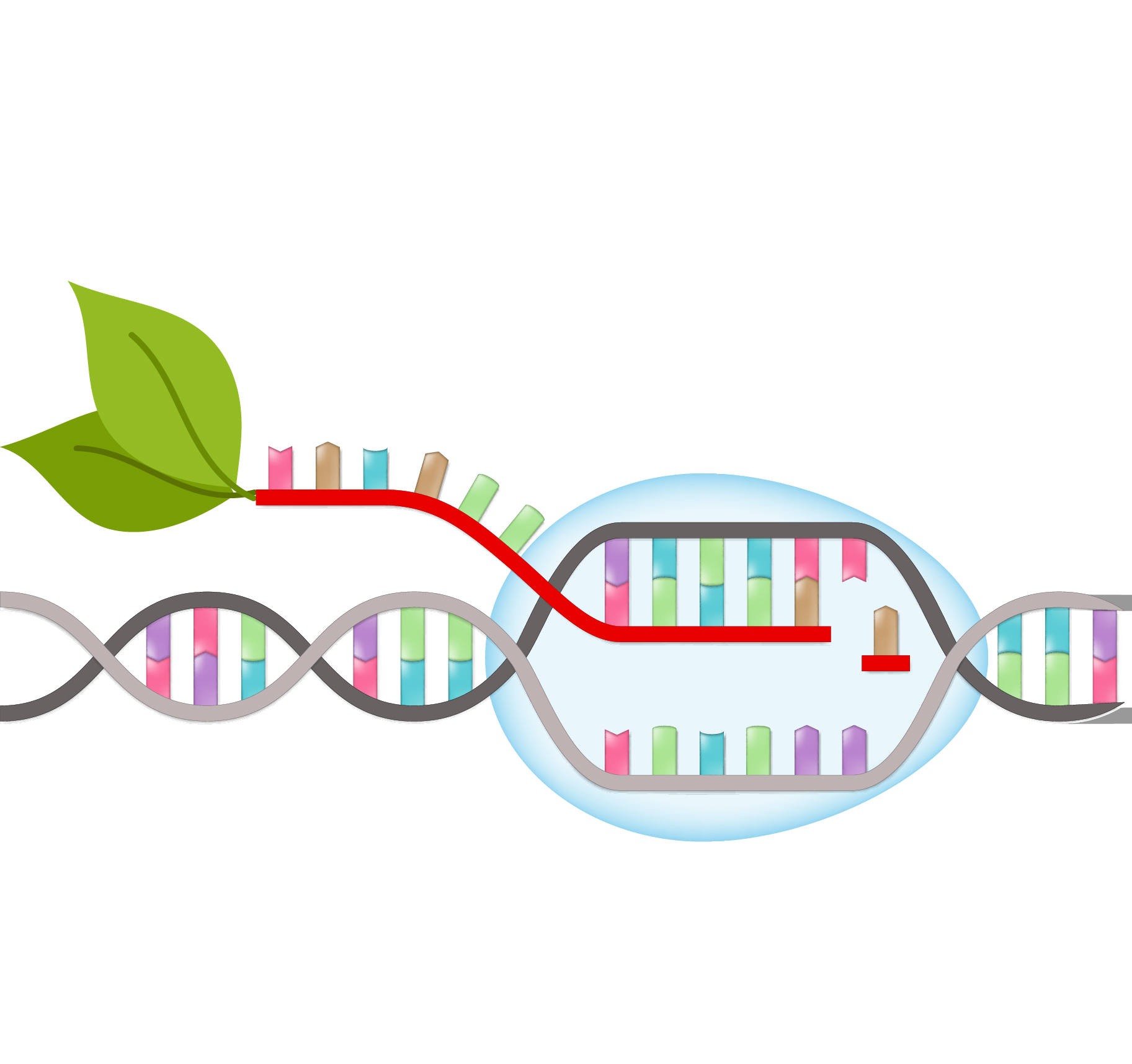
Basic properties of plant transcription
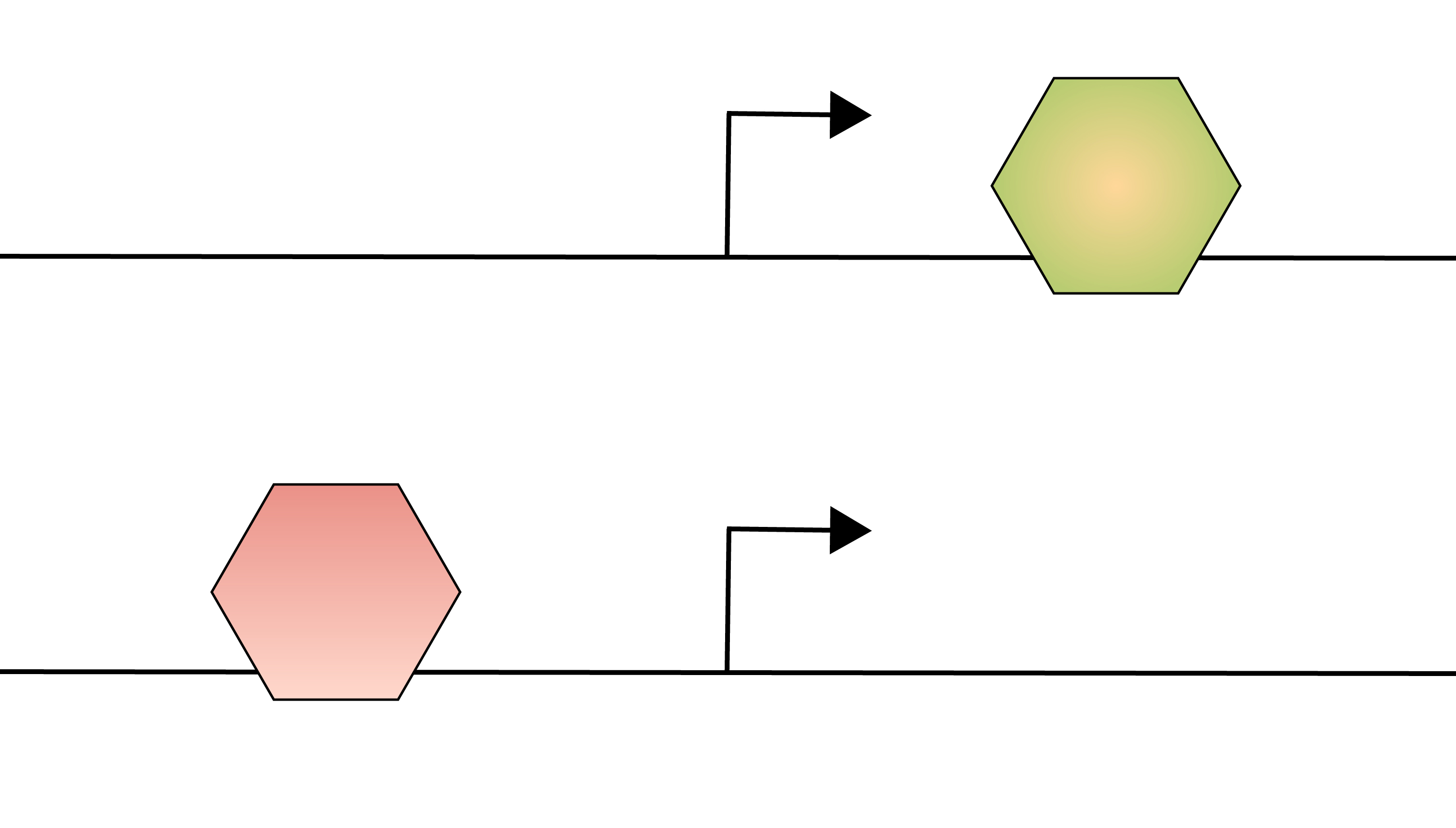
How does genomic context shape transcription factor function?

Common and divergent paths in eukaryotic transcription