We are studying the roles of protein tyrosine phosphatases (PTPs) in osteoclast (OCL) biology. PTPs are a large family of enzymes that dephosphorylate tyrosine residues in proteins, thereby regulating protein phosphorylation in concert with the protein tyrosine kinase (PTKs).
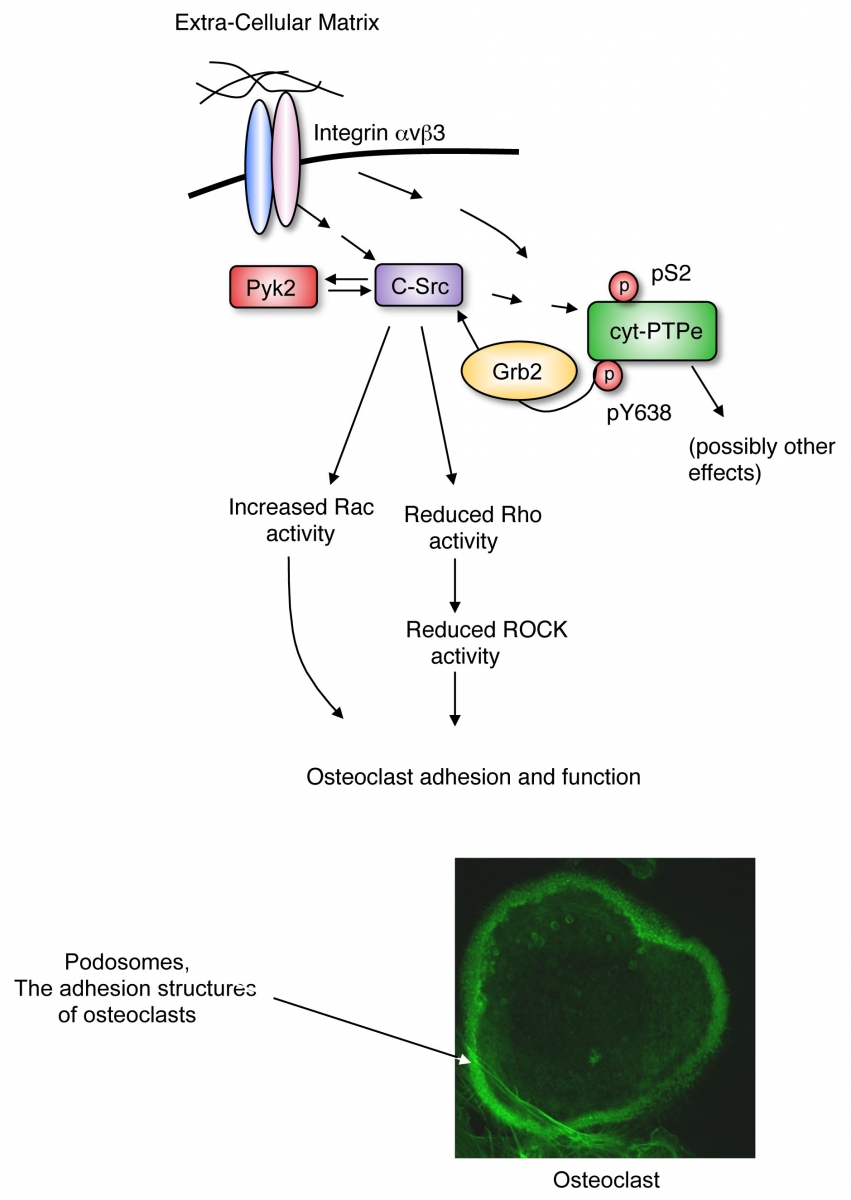
Among our studies we have shown that the non-receptor isoform of protein tyrosine phosphatase epsilon (cyt-PTPe) supports the adhesion and activity of these cells. Lack of cyt-PTPe causes structural defects in podosomes, the subcellular structures by which osteoclasts adhere to bone. At the molecular level, physical contact of OCLs with bone matrix activates signaling by the integrin receptor molecules, the mechano-sensory receptors of the cell. Activation of integrin signaling in OCLs induces phosphorylation of cyt-PTPe at its C-terminal Y638. This phosphorylation event allows the adaptor molecule Grb2 to bind cyt-PTPe and to recruit the PTK Src to its vicinity. cyt-PTPe then dephosphorylates Src at its inhibitory Y527, thus activating the kinase. Proper Src kinase activity is essential for OCLs to adhere to bone and to degrade it properly. Lack of cyt-PTPe in PTPe-deficient mice reduces Src activity in OCLs and hence harms the function of these cells, both in culture and in vivo.
Additional studies have revealed that, surprisingly, the closely-related PTP Alpha (RPTPa) does not play a unique role in osteoclasts. This is because the isoform of PTPe that is expressed in osteoclasts is the non-receptor, predominantly cytosolic form of PTPe, while PTP Alpha is expressed in these cells as a receptor-type PTP that is present at the cell membrane. Removing the PTP Alpha catalytic domains from their association with the membrane and transferring them into the cytosol allows them to function in OCLs similar to cyt-PTPe.
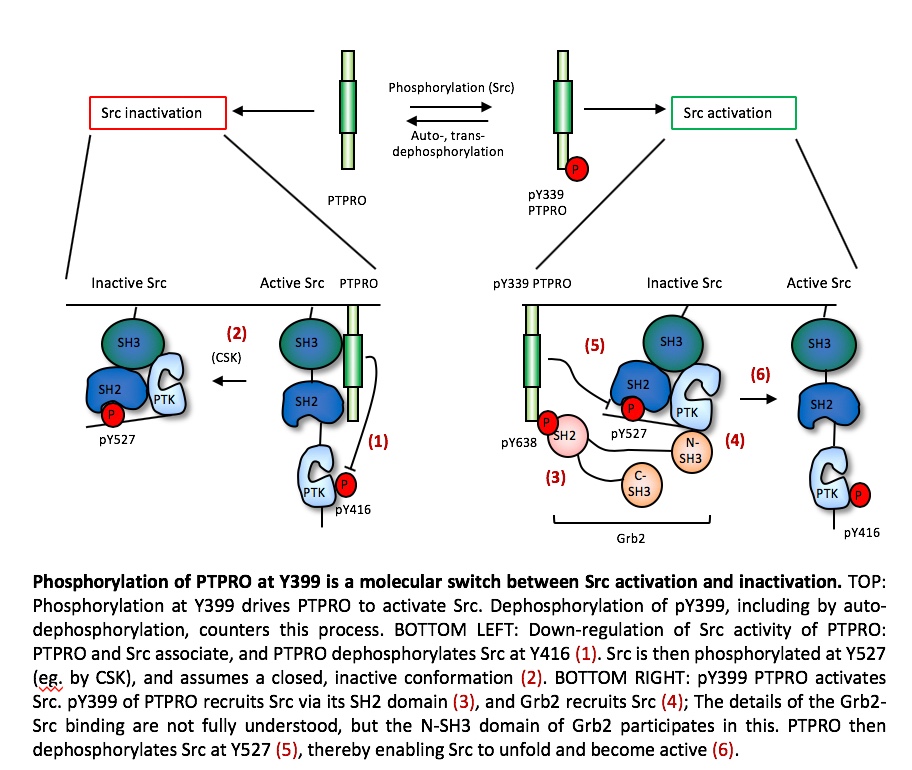
Our studies of the receptor-type PTPROt have shown that this PTP regulates Src in a more complex manner. PTPROt undergoes phosphorylation at its C-terminal Y399, which enables it to activate Src by dephosphorylating Src itself at Y527, which activates Src. In contrast, when PTPROt is not phosphorylated at Y399, this PTP down-regulates Src activity. Phosphorylation of PTPROt at Y399 is thus a molecular switch that balances between its activity as an activator vs. an inhibitor of Src, in vitro and in vivo. Accordingly, mice that completely lack PTPROt do not exhibit bone or osteoclast phenotypes since both the activation and the inhibitory functions of PTPROt towards Src are disrupted, resulting in no net change in Src or OCL activities. On the other hand, mice homozygous for Y399F PTPROt, in which the Y399 phosphorylation site has been destroyed (thereby mimicking the non-phosphorylated state of PTPROt), exhibit reduced Src activity, reduced OCL activity, and increased bone mass.
We have also studied an additional receptor-type PTP, PTPRJ (also known as DEP-1). We have shown that OCLs from mice lacking PTPRJ exhibit OCLs that do not mature properly and as a result function less well than wild-type OCLs. As a result, PTPRJ-knockout mice exhibit an increase in bone mass. Mice in which PTPRJ is removed in an OCL-specific manner behave in a similar manner, indicating that these effects are cell-autonomous in OCLs and are not caused by endocrine or other systemic effects in the mice. At the molecular level, we have shown that loss of PTPRJ in OCLs reduces the amounts of NFATc1, the key transcription factor that drives osteoclastogenesis. NFATc1 can be marked for degradation following its ubiquitination by Cbl, which can be induced by M-CSF receptor signaling. In the absence of PTPRJ M-CSF receptor signaling and Cbl activity are increased, leading to increased ubiquitination and degradation of NFATc1 and to consequent reduced OCL maturation and activity.