Leskes Lab

Research
Our research is motivated by questions in materials science for which we develop and use advanced characterization tools based on magnetic resonance.
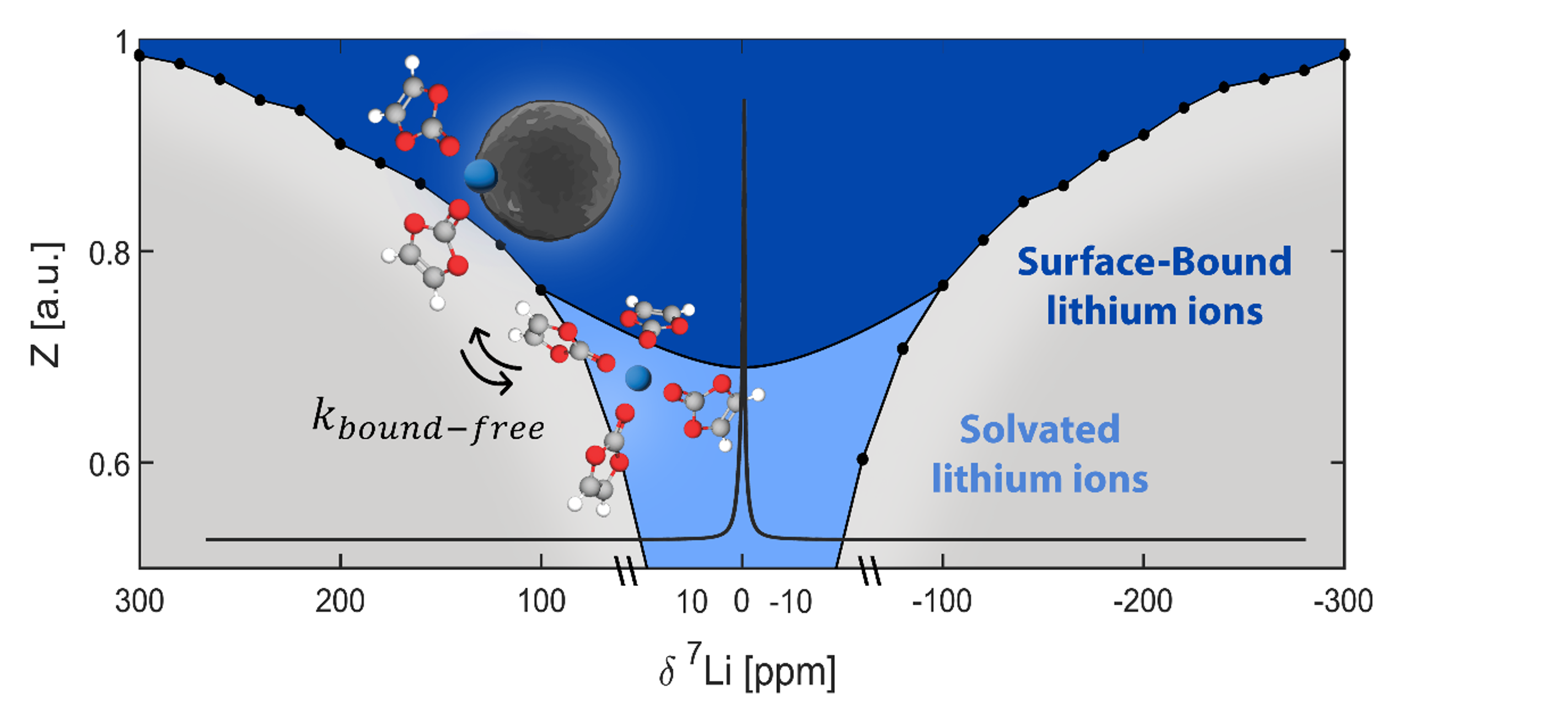
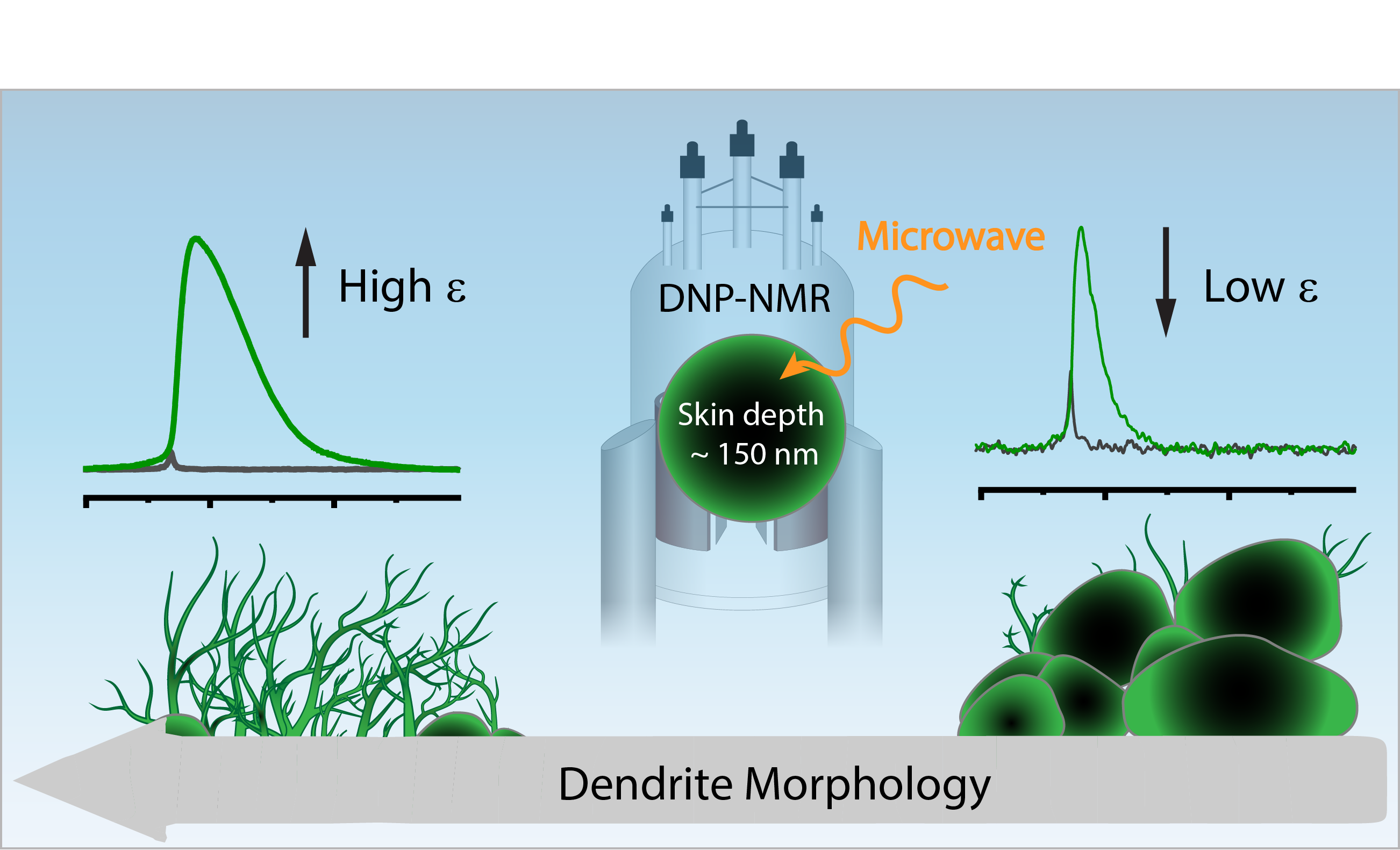
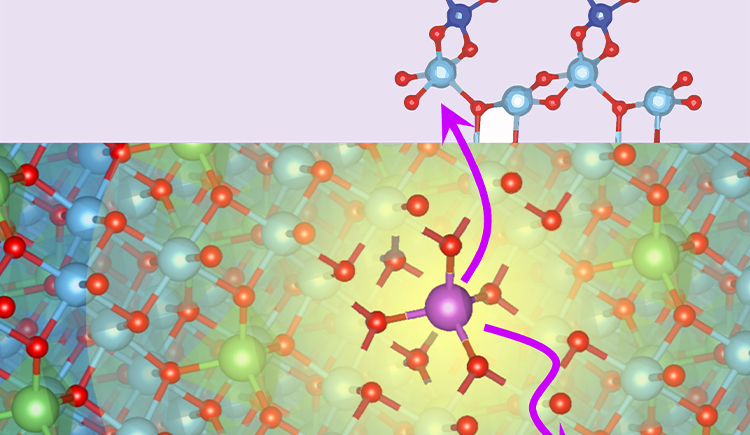
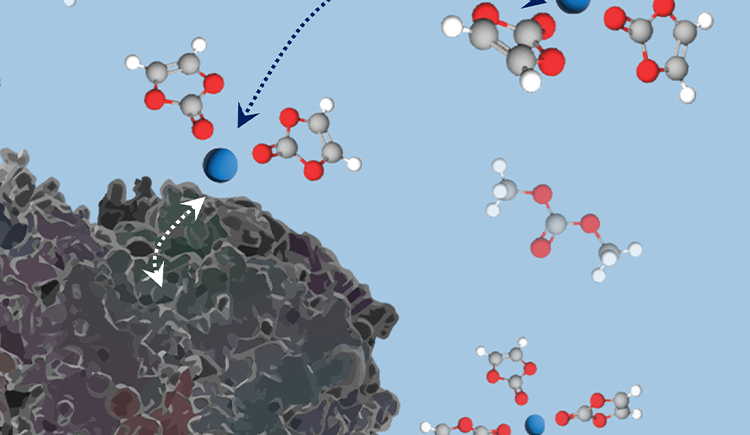
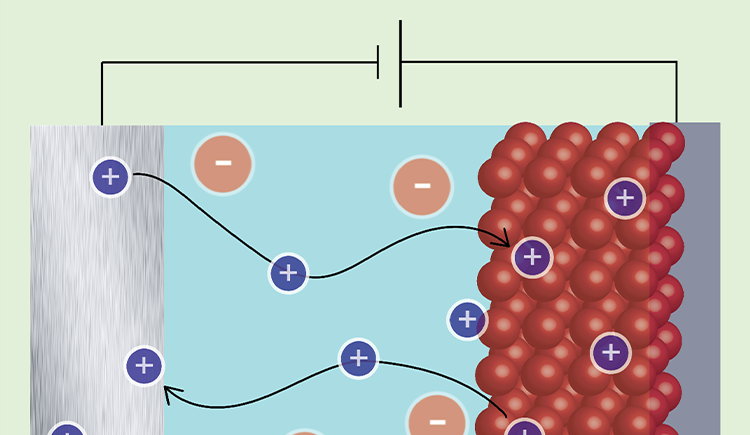
We aim to understand how the composition of materials affects their function and how we can control their function through chemical modifications. In particular we are interested in materials for energy storage and conversion and in the effect of interfacial chemistry on the functionality of electrode and electrolyte materials.
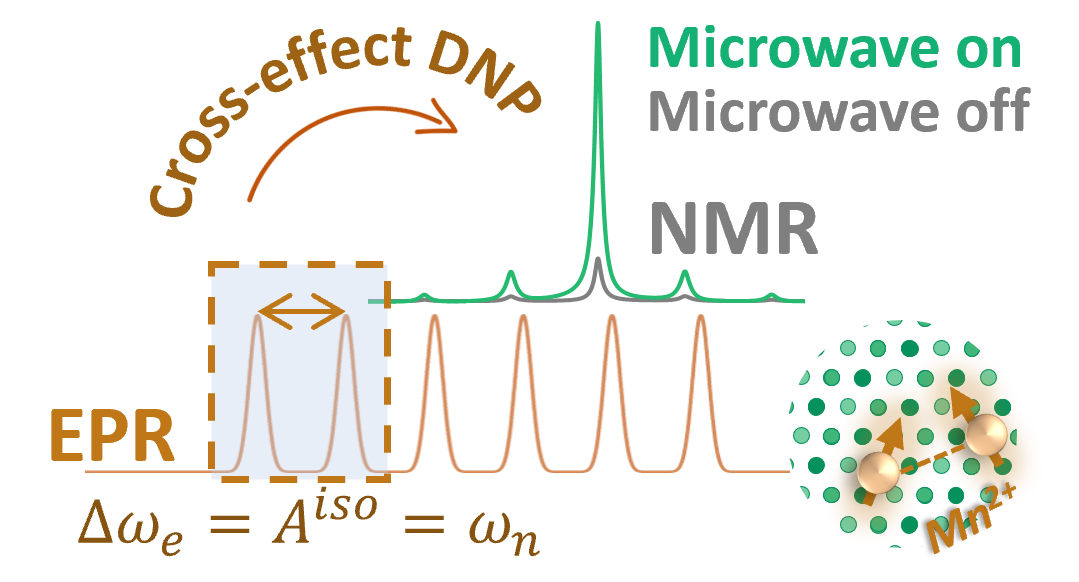
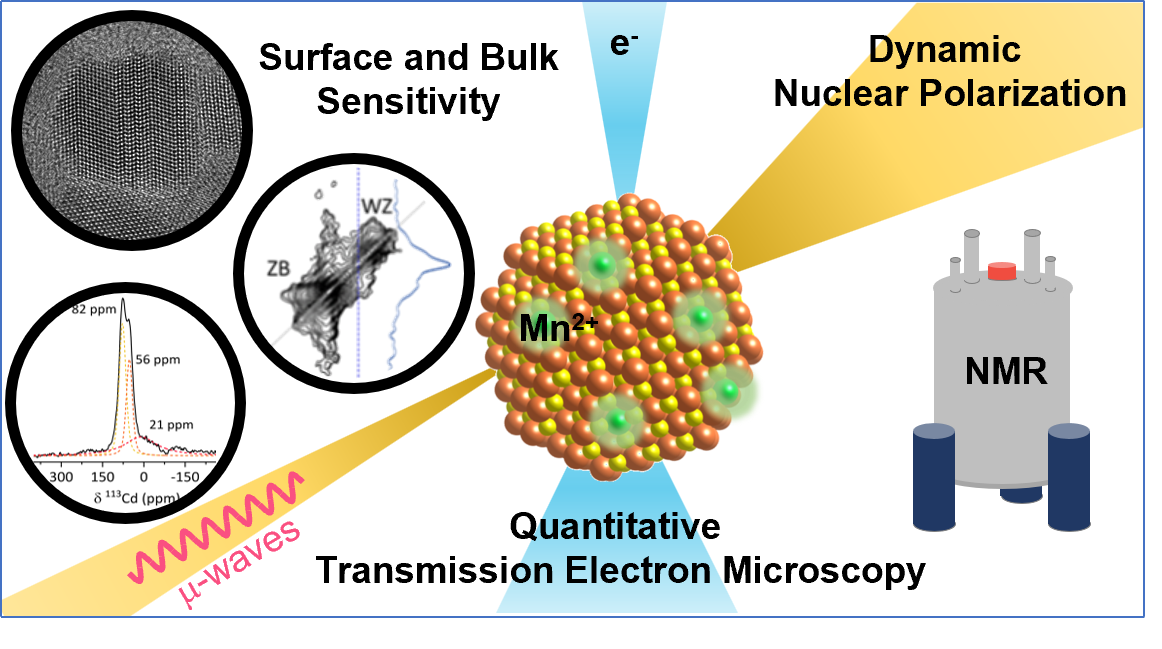
To answer these questions we use a wide range of magnetic resonance techniques: solid state NMR, Electron Paramagnetic Resonance (EPR) and Dynamic Nuclear Polarization (DNP). NMR and EPR provide insight into short range order, chemical bonding and composition and allow probing dynamic processes in a wide range of time-scales. DNP is used to boost the sensitivity of ssNMR measurements and can provide selectivity to the surface.
In addition we employ common materials science characterization tools, including electron microscopy, X-ray diffraction and X-ray photoelectron spectroscopy.
News