For many years people have asked what is the impact of DNA methylation changes in cancer? Silencing key genes? Or just responding to the true driving genomic mutations? Is it cause or consequence of gene expression?
We profiled the DNA methylation of 1538 breast tumors and 244 normal breast tissues from the METABRIC cohort, integrating genomic, transcriptional and prognostic data and generated a unified model for cancer DNA methylation that reconciles decades of conflicting evidence on passenger and driver roles for epigenetic changes in cancer.
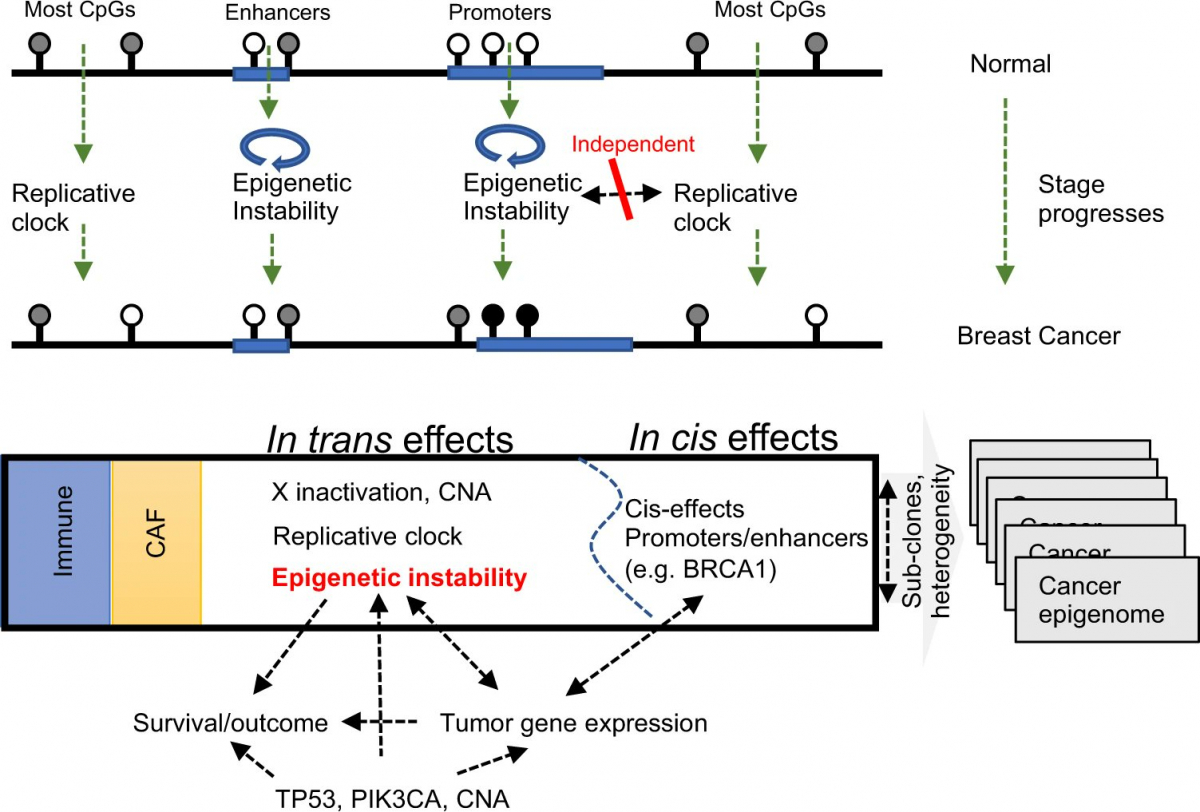
In a nutshell – as carcinogenesis progresses, tumor epigenomes are affected by widespread genome replication-dependent methylation loss, and by a second, uncorrelated epigenetic instability process modulating methylation in promoters and enhancers. These processes are each affecting a large number of genomic loci through one common mechanism (in-trans effects). Additional localized tumor-specific methylation aberrations may regulate gene expression in cis. Such cis regulation is almost always spurious (analogous to passenger genetic mutations), but in some important cases it can affect key cancer driver genes (e.g BRCA1).
We continue to investigate the role of epigenetic instability in carcinogenesis and in normal tissues, looking at additional cancer models and zooming in to the single cell level using novel protocols developed in the lab.