Vendramin R., Fu H., Fernandez Patel S., Zhao Y., Qian D., Ligammari L., Bartok O., Greenberg P., Levy R., Castro A., Thakkar K., Murai J., Lu W. T., Sng C. C., Weller C., Beattie G., Bhamra A., Farriol-Duran R., Karagianni D. & Samuels Y.
(2026)
Immunity.
SummaryDNA mutations are a well-characterized source of neoepitopes in immunotherapy. Here, we examined the contribution of dysregulated RNA processing to neoantigen production. Leveraging multi-omics and checkpoint inhibitor (CPI) response data from >1,000 patients, we identified reduced activity of the nonsense-mediated mRNA decay (NMD) pathway kinase SMG1 as a predictor of improved CPI response. NMD inhibition through SMG1 targeting stabilized transcripts containing premature termination codons, most of which were of non-mutational origin. This reshaped the major histocompatibility complex class I (MHC class I)-bound immunopeptidome and increased neoantigen abundance to levels comparable to high mutation burden tumors. Functionally, NMD inhibition drove antigen-dependent T cell-mediated tumor cell killing in vitro, promoted activation of tissue-resident T cells in patient-derived models ex vivo, and improved CPI efficacy in vivo. Our findings establish NMD inhibition as a strategy to harness a previously inaccessible source of canonical and non-canonical neoantigens, with the potential to increase tumor immunogenicity across cancers.
Marine J. C., Bartok O., Sagie S., Vitiello P. P., Bardelli A., Weller C., Chang T. G., Tonelli C., Spranger S., Ruppin E. & Samuels Y.
(2026)
Cell.
189,
8,
p. 2416-2440
Intratumor heterogeneity (ITH) encompasses genetic, epigenetic, transcriptional, proteomic, and immunopeptidomic diversity. Beyond genetic heterogeneity, it is increasingly clear that non-mutational heterogeneity and plasticity generate dynamic cancer cell states with distinct immune visibility. These layers of complexity converge on the immunopeptidome, the repertoire of peptides displayed by major histocompatibility complex molecules through which tumor cells are surveyed by T cells. Variation in antigen processing, presentation, and peptide abundance across cancer clones and cell states yields spatially and temporally distinct immunological niches that shape immune recognition and therapeutic response. Here, we summarize how multidimensional ITH manifests across cancer types and constrains immunotherapy efficacy. We propose that integrating measurements across layers is a promising direction for improving biomarker identification and informing more precise immune-based treatment strategies.
Holiar V., Rudenko V., Weller C., Naumova M., Lebon S., Canella M., Busko P., Sarusi-Portuguez A., Shalit T., Habshush Menachem A., Adir I., Petrover Z., Greenberg P., Katina C., Gradchenko P., Toval B., Yissachar N., Sagi I., Tzahor E., Levin Y., Samuels Y. & Biton M.
(2026)
BioRxiv.
The intestinal epithelium plays a pivotal role in balancing immune tolerance and inflammation, yet how it communicates tissue state to the adaptive immune system remains unclear. Here, we show that intestinal epithelial cells (IECs) encode tissue identity and injury into the major histocompatibility complex class II (MHC II) ligandome. We employed integrated single cell transcriptomics, quantitative proteomics, and high-depth in vivo immunopeptidomics to map the MHC class II self-peptidome of the mouse small intestine across epithelial and immune compartments. Mature enterocytes and intestinal stem cells (ISCs) emerged as the dominant epithelial antigen-presenting cells (APCs), displaying a compartmentalized repertoire of endogenous self-immunopeptides reflecting epithelial differentiation and function. Disruption of epithelial MHC II expression led to loss of antigenic compartmentalization, immune infiltration, extracellular matrix remodeling, and emergence of inflammation-associated immune ligands, demonstrating that epithelial MHC II is required to maintain homeostasis. Functionally, a subset of ISC-derived self-immunopeptides preferentially promotes regulatory CD4□ T cell responses, linking epithelial antigen presentation and peripheral tolerance. During gut inflammation, the epithelial MHC II landscape shifted toward damage-associated antigens. Together, these findings establish epithelial MHC II presentation as a context-dependent tissue-immune communication system that promotes tolerance in homeostasis and alerts to tissue injury during inflammation.Competing Interest StatementThe authors have declared no competing interest.Center for New Scientists at the Weizmann Institute of ScienceIsrael Science Foundation, 1587/20, 3775/25Helen and Martin Kimmel Institute for Stem Cell Research at The Weizmann Institute of ScienceMoross Integrated Cancer CenterIsrael Ministry of Science, 4631Dr. Gilbert S. Omenn and Martha A. Darling Weizmann Institute - Schneider Hospital Fund for Clinical Breakthroughs through Scientific CollaborationsSnider FoundationAbisch-Frenkel RNA Therapeutics CenterShimon and Golde PickerHerbert K. Bennett Charitable FundDwek Institute for Cancer Therapy Research
Gumpert N., Sagie S., Arnedo-Pac C., Babu T., Weller C., Gonzalez-Perez A., Wang Y., Todó L. M., Levy R., Chen X., Greenberg P., Dayan-Rubinov M., Yakubovich E., Wasserman-Bartov T., Zerbib M., Gong J., Rebernick R. J., Tercero A. O., Muriel L. A., Benedek G., Kedmi M., Oren R., Ben-Dor S., Levin Y., Troyanskaya O. G., Munzur A. D., Wyatt A. W., Cieslik M. P., Quigley D. A., Van Allen E. M., Anandasabapathy N., Mateo J., Yang X., Martínez-Jiménez F., Lopez-Bigas N. & Samuels Y.
(2026)
Cancer Discovery.
16,
2,
p. OF1-OF20
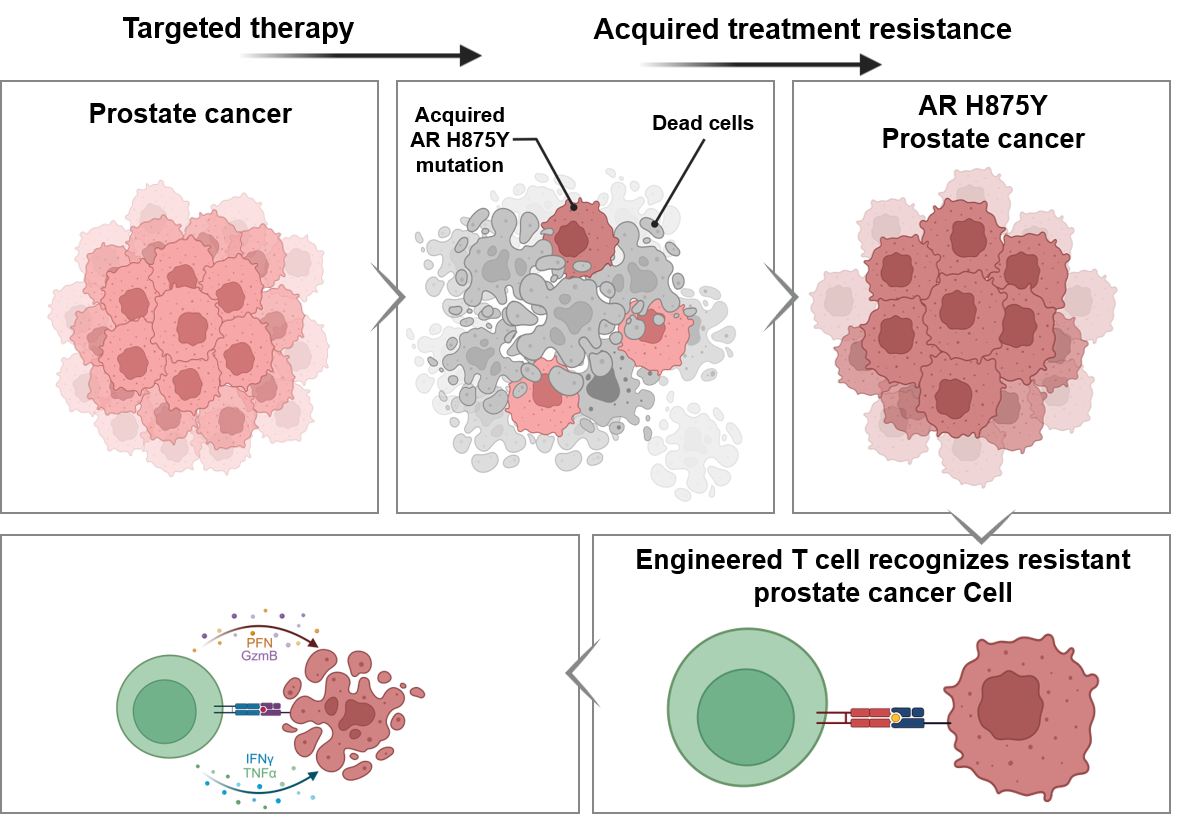
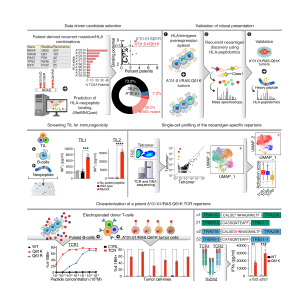
New approaches that generate long-lasting therapeutic responses in patients with therapy-resistant metastatic cancer are urgently needed. To address this challenge, we developed Spot Neoantigens in Metastases (SpotNeoMet), a novel data-driven pipeline that systematically identifies recurrently presented neopeptides in treatment-resistant patients. We identified seven therapy resistance mutations predicted to produce neopeptides presented by common HLAs. Using HLA immunopeptidomics, we discovered three novel neopeptides derived from androgen receptor (AR) H875Y, a common metastatic castration-resistant prostate cancer (mCRPC) mutation. We validated these neoantigens as highly immunogenic and then isolated and characterized cognate T-cell receptors (TCR) from healthy donor peripheral blood mononuclear cells. We demonstrated that AR H875Y-specific TCRs are highly specific and kill prostate cancer cells presenting AR neopeptides in vitro and in vivo. Our new pipeline identifies novel immunotherapy targets and potential treatment options for patients with mCRPC. Moreover, SpotNeoMet offers a systematic route to identify "HLA-peptide" pairs and their cognate TCRs across treatment-resistant cancers.Significance: As the emergence of resistance to targeted treatments in patients with metastatic cancer, there is an urgent need for innovative therapeutic approaches for this population. Our study provides a new analytic framework to identify neoantigens from treatment-resistant mutations and a proof-of-concept T cell-based immunotherapy treatment for mCRPC.
Sagie S., Babu T., Weller C., Tabachnik C., Livneh I., Quast N. P., Gumpert N., Shomuradova A., Raybould M. I., Levy R., Malko D., Alfia A., Ben Lulu T., Alon M., Herrera F., Kutuzov M., Zerbib M., Greenberg P., Wasserman-Bartov T., Benedek G., Levin Y., Stossel C., Kamer I., Golan T., Oren R., Shmueli M., Bartok O., Bar J., Cohen J., Dushek O., Deane C. M. & Samuels Y.
(2025)
Cell Reports Medicine.
6,
12,
102506.
Adoptive cell therapy (ACT) targeting tumor-specific antigens holds promise for solid tumors, but limited neoantigen presentation remains a key barrier to efficacy. Here, we identify and characterize a T cell receptor (TCR), T104, for the KRAS.G12V mutation, a prevalent neoantigen in colorectal, lung, and pancreatic cancers. TCR-T104 selectively recognizes and kills KRAS.G12V-expressing tumor cells. Combining T cell therapy with lymphodepleting chemotherapy significantly enhances tumor cell killing, particularly by TCR-T cells, tumor-infiltrating lymphocytes (TILs), and T cell engager antibodies across multiple cancer types and target antigens. Mechanistically, chemotherapy upregulates immunoproteasome activity and human leukocyte antigen (HLA)-I surface expression. HLA-immunopeptidome analyses reveal that chemotherapy remodels the antigenic landscape across tumor cell lines and in vivo models, increasing peptide abundance and hydrophobicity while altering proteasomal cleavage preferences. These findings establish a synergistic role for chemotherapy in enhancing neoantigen presentation and T cell-mediated tumor recognition and suggest that fine-tuning these regimens could improve ACT efficacy, particularly in tumors with low-abundance neoantigens.
Chemla Y., Itzhaki O., Melamed S., Weller C., Sade Y., Manich P., Reshef K., Xenidis N., Maliah A., Levy G., Parikh R., Bartok O., Levy O., Tal I., Aziel G., Nissani A., Yunger S., Likonen D., Kliminsky V., Golan T., Capron C., Ace V., Levy R., Rasoulouniriana D., Eyal Z., Barzilay Y., Balaban R., Khateeb A., Khosravi R., Grau A., Ziv T., Greenberg P., Netanely D., Vaknin H., Wu X., Amitay Y., Brenner R., Martínez Gómez J. M., Hershkovitz D., Yardeni T., Zemser-Werner V., Kobiler O., Friedmann Y., Bassan D., Shamir R., Eisenbach L., Santana-Magal N., Milyavsky M., Eisenberg G., Keren L., Cohen M., Gur D., Barak B., Lotem M., Sprinzak D., Greenberger S., Fisher D., Besser M. J., Khaled M., Close P., Shapira R., Apcher S., Madi A., Levesque M. P., Rapino F., Carmi Y., Parikh S., Samuels Y. & Levy C.
(2025)
Cell.
189,
1,
p. 233-251.e29
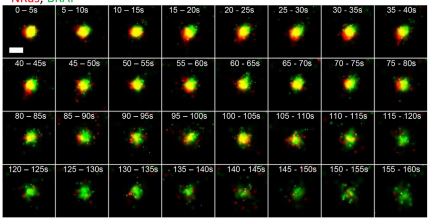
While melanoma cells often express a high burden of mutated proteins, the infiltration of reactive T cells rarely results in tumor-eradicating immunity. We discovered that large extracellular vesicles, known as melanosomes, secreted by melanoma cells are decorated with major histocompatibility complex (MHC) molecules that stimulate CD8+ T cells through their T cell receptor (TCR), causing T cell dysfunction and apoptosis. Immunopeptidomic and T cell receptor sequencing (TCR-seq) analyses revealed that these melanosomes carry MHC-bound tumor-associated antigens with higher affinity and immunogenicity, which compete with their tumor cell of origin for direct TCR-MHC interactions. Analysis of biopsies from melanoma patients confirmed that melanosomes trap infiltrating lymphocytes, induce partial activation, and decrease CD8+ T cell cytotoxicity. Inhibition of melanosome secretion in vivo significantly reduced tumor immune evasion. These findings suggest that MHC export protects melanoma from the cytotoxic effects of T cells. Our study highlights a novel immune evasion mechanism and proposes a therapeutic avenue to enhance tumor immunity.
Weller C., Bartok O., McGinnis C. S., Palashati H., Chang T. G., Malko D., Shmueli M. D., Nagao A., Hayoun D., Murayama A., Sakaguchi Y., Poulis P., Khatib A., Erlanger Avigdor B., Gordon S., Cohen Shvefel S., Zemanek M. J., Nielsen M. M., Boura-Halfon S., Sagie S., Gumpert N., Yang W., Alexeev D., Kyriakidou P., Yao W., Zerbib M., Greenberg P., Benedek G., Litchfield K., Petrovich-Kopitman E., Nagler A., Oren R., Ben-Dor S., Levin Y., Pilpel Y., Rodnina M., Cox J., Merbl Y., Satpathy A. T., Carmi Y., Erhard F., Suzuki T., Buskirk A. R., Olweus J., Ruppin E., Schlosser A. & Samuels Y.
(2025)
Cancer Cell.
43,
5,
p. 823-840.e18
Aberrant peptides presented by major histocompatibility complex (MHC) molecules are targets for tumor eradication, as these peptides can be recognized as foreign by T cells. Protein synthesis in malignant cells is dysregulated, which may result in the generation and presentation of aberrant peptides that can be exploited for T cell-based therapies. To investigate the role of translational dysregulation in immunological tumor control, we disrupt translation fidelity by deleting tRNA wybutosine (yW)-synthesizing protein 2 (TYW2) in tumor cells and characterize the downstream impact on translation fidelity and immunogenicity using immunopeptidomics, genomics, and functional assays. These analyses reveal that TYW2 knockout (KO) cells generate immunogenic out-of-frame peptides. Furthermore, Tyw2 loss increases tumor immunogenicity and leads to anti-programmed cell death 1 (PD-1) checkpoint blockade sensitivity in vivo. Importantly, reduced TYW2 expression is associated with increased response to checkpoint blockade in patients. Together, we demonstrate that defects in translation fidelity drive tumor immunogenicity and may be leveraged for cancer immunotherapy.
Shvefel S. C., Pai J. A., Cao Y., Pal L. R., Bartok O., Levy R., Zemanek M. J., Weller C., Herzog E., Yao W., Hiam-Galvez K. J., Cheng K., Yin Y., Du P. P., Raposo C. J., Gumpert N., Welti M., Martínez Gómez J. M., Sella F., Yakubovich E., Orr I., Ben-Dor S., Oren R., Fellus-Alyagor L., Golani O., Brenner O. J., Salame T. M., Zerbib M., Goliand I., Ranmar D., Savchenko I., Ketrarou N., Schäffer A. A., Dahan R., Levesque M. P., Ruppin E., Satpathy A. T. & Samuels Y.
(2025)
Cancer Discovery.
15,
3,
p. 553-577
Low intratumor heterogeneity correlates with increased patient survival and immunotherapy response. However, even highly homogeneous tumors are vari-ably aggressive, and the immunologic factors impacting aggressiveness remain understudied. In this study, we analyzed the mechanisms underlying immune escape in murine tumors with low intratumor heterogeneity. We used immunophenotyping and single-cell RNA sequencing to compare the temporal growth of in vivo transplanted, genetically similar, rejected and nonrejected single-cell clones. Nonrejected clones showed high infiltration of tumor-associated macrophages, lower T cell infiltra-tion, and increased T cell exhaustion when compared with rejected clones. Comparative analysis of rejection-associated gene expression programs, combined with in vivo CRISPR knockout screens of candidate regulators, identified macrophage migration inhibitory factor (Mif) as a major con-tributor to preventing immune rejection. Mif knockout resulted in smaller tumors and reduced tumor-associated macrophage infiltration. These results were validated in patients with melanoma. Overall, our homogeneous tumor system can uncover factors regulating growth variability and identifies Mif as critical in aggressive melanoma. Significance: In this study, we find that Mif expression is associated with tumor growth and aggressiveness, specifically in tumors with low heterogeneity. These findings could facilitate the development of new strategies to treat patients with homogeneous, high MIFexpressing tumors that are unresponsive to immune checkpoint therapy.
OBrien H., Salm M., Morton L. T., Szukszto M., OFarrell F., Boulton C., King L., Bola S. K., Becker P. D., Craig A., Nielsen M., Samuels Y., Swanton C., Mansour M. R., Hadrup S. R. & Quezada S. A.
(2024)
PLoS Computational Biology.
20,
11,
e1012511.
Neoantigen immunogenicity prediction is a highly challenging problem in the development of personalised medicines. Low reactivity rates in called neoantigens result in a difficult prediction scenario with limited training datasets. Here we describe ImmugenX, a modular protein language modelling approach to immunogenicity prediction for CD8+ reactive epitopes. ImmugenX comprises of a pMHC encoding module trained on three pMHC prediction tasks, an optional TCR encoding module and a set of context specific immunogenicity prediction head modules. Compared with state-of-the-art models for each task, ImmugenXs encoding module performs comparably or better on pMHC binding affinity, eluted ligand prediction and stability tasks. ImmugenX outperforms all compared models on pMHC immunogenicity prediction (Area under the receiver operating characteristic curve = 0.619, average precision: 0.514), with a 7% increase in average precision compared to the next best model. ImmugenX shows further improved performance on immunogenicity prediction with the integration of TCR context information. ImmugenX performance is further analysed for interpret-ability, which locates areas of weakness found across existing immunogenicity models and highlight possible biases in public datasets.
OBrien H., Salm M., Morton L., Szukszto M., OFarrell F., Boulton C., Becker P. D., Samuels Y., Swanton C., Mansour M. R., Reker Hadrup S. & Quezada S. A.
(2023)
Nature Reviews Cancer.
4,
12,
p. 1618-1621
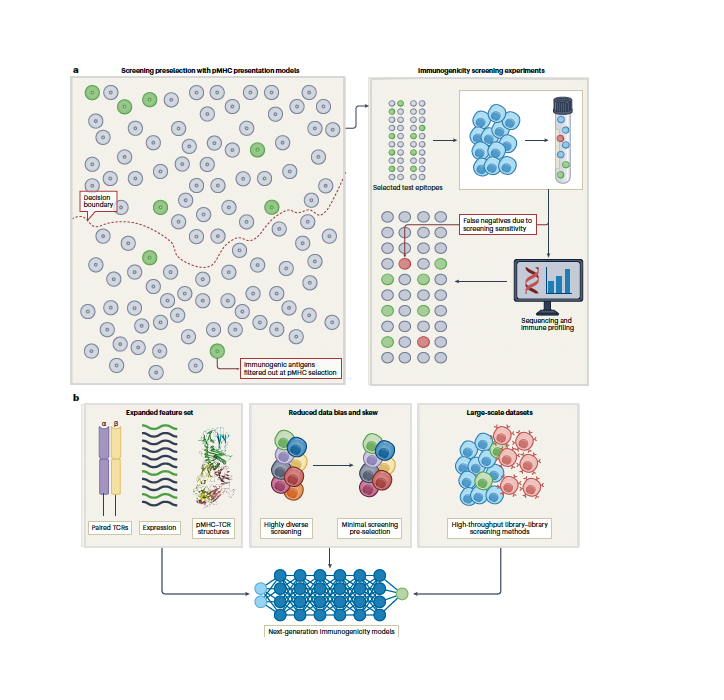
Neoantigen immunogenicity prediction is a burgeoning field with vast potential; however, the shortage of high-quality data and biases in current datasets limit model generalizability. Here we discuss some of the pitfalls that may underly this limited performance and propose a path forward.
Traditional immunotherapies provide clinical benefits to only a few patients with solid tumors, highlighting the urgent need for more effective approaches. Traditional immunotherapies rely on the presentation of cancer antigens, with neoantigens being highly important in this context as they are specific to malignant tissue but not healthy tissue. The quantity of neoantigens is often associated with clinical benefit, but it cannot fully explain or predict patient response. In this Viewpoint, we highlight several qualitative aspects that should be considered in neoantigen-based therapy. We emphasize the distinction between private and recurrent neoantigens, discuss the importance of neoantigen clonality, and describe new subtypes of neopeptides that further diversify the potential of neoantigens in immunotherapy.
The remarkable capacity of immunotherapies to induce durable regression in some patients with metastatic cancer relies heavily on T cell recognition of tumor-presented antigens. As checkpoint-blockade therapy has limited efficacy, tumor antigens have the potential to be exploited for complementary treatments, many of which are already in clinical trials. The surge of interest in this topic has led to the expansion of the tumor antigen landscape with the emergence of new antigen categories. Nonetheless, how different antigens compare in their ability to elicit efficient and safe clinical responses remains largely unknown. Here, we review known cancer peptide antigens, their attributes and the relevant clinical data and discuss future directions.
Javitt A., Shmueli M. D., Kramer M. P., Kolodziejczyk A. A., Cohen I. J., Radomir L., Sheban D., Kamer I., Litchfield K., Bab-Dinitz E., Zadok O., Neiens V., Ulman A., Wolf-Levy H., Eisenberg-Lerner A., Kacen A., Alon M., Rêgo A. T., Stacher-Priehse E., Lindner M., Koch I., Bar J., Swanton C., Samuels Y., Levin Y., da Fonseca P. C., Elinav E., Friedman N., Meiners S. & Merbl Y.
(2023)
Nature Reviews Cancer.
4,
5,
p. 629-647
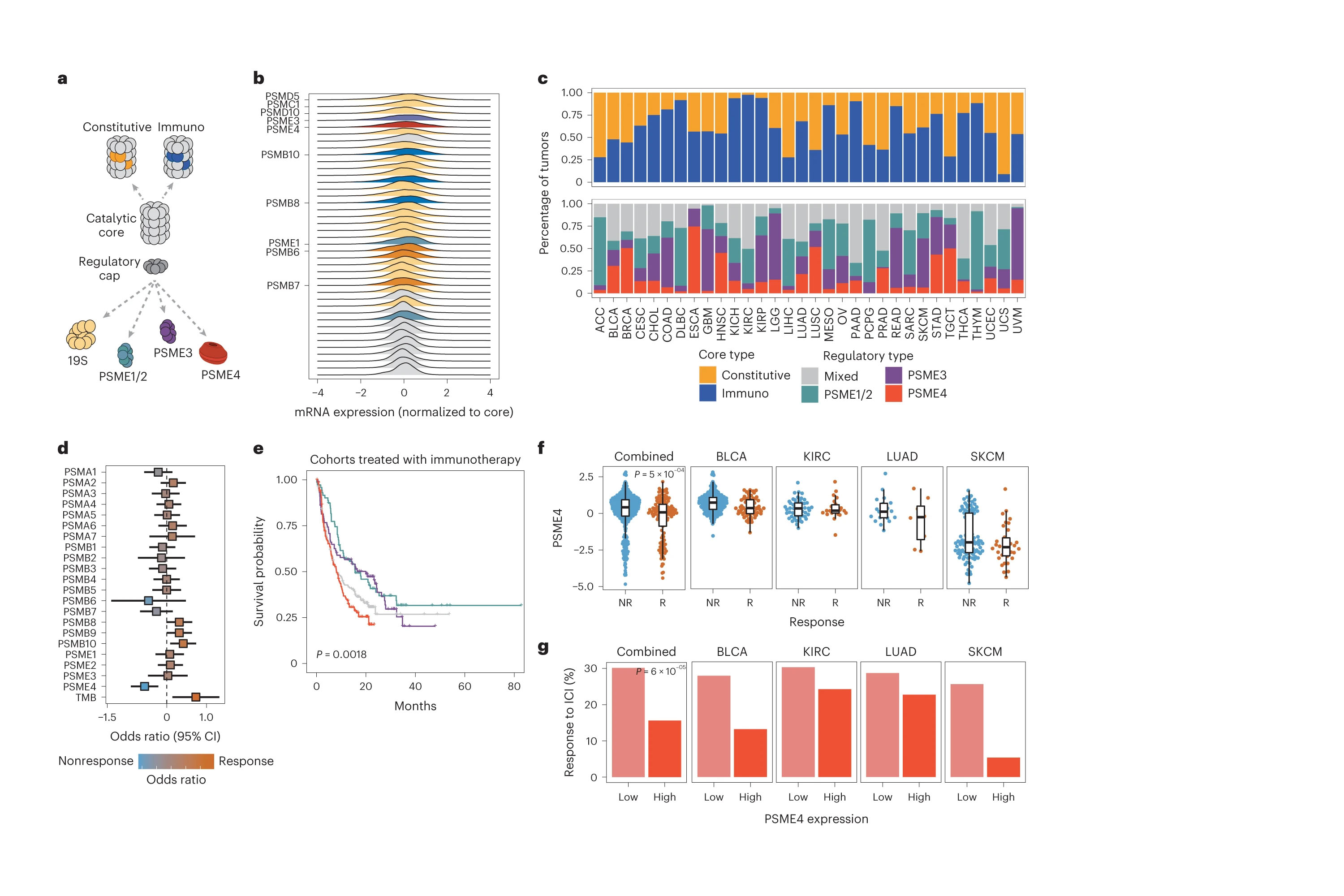
Immunotherapy revolutionized treatment options in cancer, yet the mechanisms underlying resistance in many patients remain poorly understood. Cellular proteasomes have been implicated in modulating antitumor immunity by regulating antigen processing, antigen presentation, inflammatory signaling and immune cell activation. However, whether and how proteasome complex heterogeneity may affect tumor progression and the response to immunotherapy has not been systematically examined. Here, we show that proteasome complex composition varies substantially across cancers and impacts tumorimmune interactions and the tumor microenvironment. Through profiling of the degradation landscape of patient-derived non-small-cell lung carcinoma samples, we find that the proteasome regulator PSME4 is upregulated in tumors, alters proteasome activity, attenuates presented antigenic diversity and associates with lack of response to immunotherapy. Collectively, our approach affords a paradigm by which proteasome composition heterogeneity and function should be examined across cancer types and targeted in the context of precision oncology.
Levy R., Alter Regev T., Paes W., Gumpert N., Cohen Shvefel S., Bartok O., Dayan-Rubinov M., Alon M., Shmueli M., Levin Y., Merbl Y., Ternette N. & Samuels Y.
(2023)
Molecular and Cellular Proteomics.
22,
4,
100519.
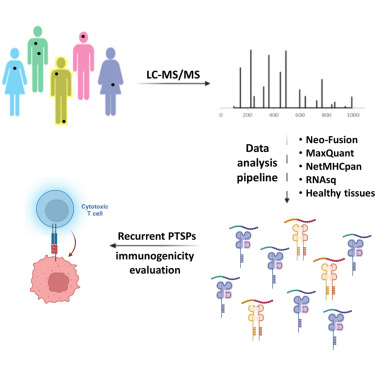
Post-translational spliced peptides (PTSPs) are a unique class of peptides that have been found to be presented by HLA-class-I molecules in cancer (1). Thus far, no consensus has been reached on the proportion of PTSPs in the immunopeptidome, with estimates ranging from 2% to as high as 45% and stirring significant debate (2-8). Furthermore, the role of the HLA-class-II pathway in PTSP presentation has been studied only in diabetes (9). Here, we exploit our large-scale cancer peptidomics database and our newly devised pipeline for filtering spliced peptide predictions to identify recurring spliced peptides, both for HLA-class-I and -II complexes. Our results indicate that HLA-class-I spliced peptides account for a low percentage of the immunopeptidome (less than 3.1%), yet are larger in number relative to other types of identified aberrant peptides. Therefore, spliced peptides significantly contribute to the repertoire of presented peptides in cancer cells. In addition, we identified HLA-class-II-bound spliced peptides, but to a lower extent (less than 0.5%). The identified spliced peptides include cancer- and immune-associated genes, such as the MITF oncogene, DAPK1 tumor suppressor and HLA-E, which were validated using synthetic peptides. The potential immunogenicity of the DAPK1- and HLA-E-derived PTSPs was also confirmed. In addition, a reanalysis of our published mouse single-cell clone immunopeptidome dataset showed that most of the spliced peptides were found repeatedly in a large number of the single-cell clones. Establishing a novel search-scheme for the discovery and evaluation of recurring PTSPs among cancer patients may assist in identifying potential novel targets for immunotherapy.
Kacen A., Javitt A., Kramer M. P., Morgenstern D., Tsaban T., Shmueli M. D., Teo G. C., Leprevost F. d. V., Barnea E., Yu F., Admon A., Eisenbach L., Samuels Y., Schueler-Furman O., Levin Y., Nesvizhskii A. & Merbl Y.
(2023)
41,
2,
p. 239-251
Post-translational modification (PTM) of antigens provides an additional source of specificities targeted by immune responses to tumors or pathogens, but identifying antigen PTMs and assessing their role in shaping the immunopeptidome is challenging. Here we describe the Protein Modification Integrated Search Engine (PROMISE), an antigen discovery pipeline that enables the analysis of 29 different PTM combinations from multiple clinical cohorts and cell lines. We expanded the antigen landscape, uncovering human leukocyte antigen class I binding motifs defined by specific PTMs with haplotype-specific binding preferences and revealing disease-specific modified targets, including thousands of new cancer-specific antigens that can be shared between patients and across cancer types. Furthermore, we uncovered a subset of modified peptides that are specific to cancer tissue and driven by post-translational changes that occurred in the tumor proteome. Our findings highlight principles of PTM-driven antigenicity, which may have broad implications for T cell-mediated therapies in cancer and beyond.
NRas is a key mediator of the mitogenic pathway in normal cells and in cancer cells. Its dynamics and nanoscale organization at the plasma membrane (PM) facilitate its signaling. Here, we used two-color photoactivated localization microscopy to resolve the organization of individual NRas and associated signaling proteins in live melanoma cells, with resolution down to ∼20 nm. Upon EGF activation, a fraction of NRas and BRAF (dis)assembled synchronously at the PM in co-clusters. NRas and BRAF clusters associated with GPI-enriched domains, serving as possible nucleation sites for these clusters. NRas and BRAF association in mutual clusters was reduced by the NRas farnesylation inhibitor lonafarnib, yet enhanced by the BRAF inhibitor vemurafenib. Surprisingly, dispersed NRas molecules associated with the periphery of self-clusters of either Grb2 or NF1. Thus, NRas-mediated signaling, which is critical in health and disease, is regulated by dynamic interactions with functional clusters of BRAF or other related proteins at the PM.
Over the last decade, it has become clear that the genomic landscapes of tumors profoundly impact their immunogenicity and how tumor cells interact with immune cells. Whereas past discoveries mainly focused on the interplay between tumor immunogenicity and tumor mutational burden (TMB), under the assumption that a higher mutation load would give rise to a better patient response to immune checkpoint blockade (ICB) therapies, we and others have underlined intratumor heterogeneity (ITH) as an important determinant of the magnitude of the anti-tumor response and the nature of the tumor microenvironment. In this Review, we define TMB vs. ITH and how the two factors are being inferred from data, examine key findings in the cancer immunogenomics literature deciphering the complex crosstalk between TMB, ITH and anti-tumor immunity in human cancers and in-vivo models, and discuss the mutual influence of ITH and immunity - how the anti-tumor response can give rise to tumors with higher ITH, and how higher ITH can put shackles on the anti-tumor response.
Melanoma, a skin cancer that develops from pigment cells, has been studied intensively, particularly in terms of the immune response to tumours, and has been used as a model for the development of immunotherapy. This is due, in part, to the high mutational burden observed in melanomas, which increases both their immunogenicity and the infiltration of immune cells into the tumours, compared with other types of cancers. The immune response to melanomas involves a complex set of components and interactions. As the tumour evolves, it accumulates an increasing number of genetic and epigenetic alterations, some of which contribute to the immunogenicity of the tumour cells and the infiltration of immune cells. However, tumour evolution also enables the development of resistance mechanisms, which, in turn, lead to tumour immune escape. Understanding the interactions between melanoma tumour cells and the immune system, and the evolving changes within the melanoma tumour cells, the immune system and the microenvironment, is essential for the development of new cancer therapies. However, current research suggests that other extrinsic factors, such as the microbiome, may play a role in the immune response to melanomas. Here, we review the mechanisms underlying the immune response in the tumour and discuss recent advances as well as strategies for treatment development.
Spencer C. N., McQuade J. L., Gopalakrishnan V., McCulloch J. A., Vetizou M., Cogdill A. P., Wadud Khan M. A., Zhang X., White M. G., Peterson C. B., Wong M. C., Morad G., Rodgers T., Badger J. H., Helmink B. A., Andrews M. C., Rodrigues R. R., Morgun A., Kim Y. S., Roszik J., Hoffman K. L., Zheng J., Zhou Y., Medik Y. B., Kahn L. M., Johnson S., Hudgens C. W., Wani K., Gaudreau P. O., Harris A. L., Jamal M. A., Baruch E. N., Perez-Guijarro E., Day C. P., Merlino G., Pazdrak B., Lochmann B. S., Szczepaniak-Sloane R. A., Arora R., Anderson J., Zobniw C. M., Posada E., Sirmans E., Simon J., Haydu L. E., Burton E. M., Wang L., Dang M., Clise-Dwyer K., Schneider S., Chapman T., Anang N. A. A., Duncan S., Toker J., Malke J. C., Glitza I. C., Amaria R. N., Tawbi H. A., Diab A., Wong M. K., Patel S. P., Woodman S. E., Davies M. A., Ross M. I., Gershenwald J. E., Lee J. E., Hwu P., Jensen V., Samuels Y., Straussman R., Ajami N. J., Nelson K. C., Nezi L., Petrosino J. F., Andrew Futreal P., Lazar A. J., Hu J., Jenq R. R., Tetzlaff M. T., Yan Y., Garrett W. S., Huttenhower C., Sharma P., Watowich S. S., Allison J. P., Cohen L., Trinchieri G., Daniel C. R. & Wargo J. A.
(2021)
Science.
374,
6575,
p. 1632-1640
Gut bacteria modulate the response to immune checkpoint blockade (ICB) treatment in cancer, but the effect of diet and supplements on this interaction is not well studied. We assessed fecal microbiota profiles, dietary habits, and commercially available probiotic supplement use in melanoma patients and performed parallel preclinical studies. Higher dietary fiber was associated with significantly improved progression-free survival in 128 patients on ICB, with the most pronounced benefit observed in patients with sufficient dietary fiber intake and no probiotic use. Findings were recapitulated in preclinical models, which demonstrated impaired treatment response to anti-programmed cell death 1 (anti-PD-1)-based therapy in mice receiving a low-fiber diet or probiotics, with a lower frequency of interferon-g-positive cytotoxic T cells in the tumor microenvironment. Together, these data have clinical implications for patients receiving ICB for cancer.
Peri A., Greenstein E., Alon M., Pai J. A., Dingjan T., Reich-Zeliger S., Barnea E., Barbolin C., Levy R., Arnedo-Pac C., Kalaora S., Dassa B., Feldmesser E., Shang P., Greenberg P., Levin Y., Benedek G., Levesque M. P., Adams D. J., Lotem M., Wilmott J. S., Scolyer R. A., Jönsson G. B., Admon A., Rosenberg S. A., Cohen C. J., Niv M. Y., Lopez-Bigas N., Satpathy A. T., Friedman N. & Samuels Y.
(2021)
Journal of Clinical Investigation.
131,
20,
e129466.
Neoantigens are now recognized drivers of the antitumor immune response. Recurrent neoantigens, shared among groups of patients, have thus become increasingly coveted therapeutic targets. Here, we report on the data-driven identification of a robustly presented, immunogenic neoantigen that is derived from the combination of HLA-A*01:01 and RAS.Q61K. Analysis of large patient cohorts indicated that this combination applies to 3% of patients with melanoma. Using HLA peptidomics, we were able to demonstrate robust endogenous presentation of the neoantigen in 10 tumor samples. We detected specific reactivity to the mutated peptide within tumor-infiltrating lymphocytes (TILs) from 2 unrelated patients, thus confirming its natural immunogenicity. We further investigated the neoantigen-specific clones and their T cell receptors (TCRs) via a combination of TCR sequencing, TCR overexpression, functional assays, and single-cell transcriptomics. Our analysis revealed a diverse repertoire of neoantigen-specific clones with both intra- and interpatient TCR similarities. Moreover, 1 dominant clone proved to cross-react with the highly prevalent RAS.Q61R variant. Transcriptome analysis revealed a high association of TCR clones with specific T cell phenotypes in response to cognate melanoma, with neoantigen-specific cells showing an activated and dysfunctional phenotype. Identification of recurrent neoantigens and their reactive TCRs can promote "off-theshelf" precision immunotherapies, alleviating limitations of personalized treatments.
Nagler A., Kalaora S., Barbolin C., Gangaev A., Ketelaars S. L., Alon M., Pai J., Benedek G., Yahalom-Ronen Y., Erez N., Greenberg P., Yagel G., Peri A., Levin Y., Satpathy A. T., Bar-Haim E., Paran N., Kvistborg P. & Samuels Y.
(2021)
Cell reports (Cambridge).
35,
13,
109305.
The human leukocyte antigen (HLA)-bound viral antigens serve as an immunological signature that can be selectively recognized by T cells. As viruses evolve by acquiring mutations, it is essential to identify a range of presented viral antigens. Using HLA peptidomics, we are able to identify severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-derived peptides presented by highly prevalent HLA class I (HLA-I) molecules by using infected cells as well as overexpression of SARS-CoV-2 genes. We find 26 HLA-I peptides and 36 HLA class II (HLA-II) peptides. Among the identified peptides, some are shared between different cells and some are derived from out-of-frame open reading frames (ORFs). Seven of these peptides were previously shown to be immunogenic, and we identify two additional immunoreactive peptides by using HLA multimer staining. These results may aid the development of the next generation of SARS-CoV-2 vaccines based on presented viral-specific antigens that span several of the viral genes. [Display omitted] HLA peptidomics enable the identification of SARS-CoV-2-derived HLA peptidesSARS-CoV-2 peptides can be derived from canonical and non-canonical ORFsShared SARS-CoV-2 peptides are identified in different cell typesSeveral SARS-CoV-2 peptides are immunogenic Using HLA peptidomics, Nagler et al. identify SARS-CoV-2-derived peptides presented by highly prevalent HLA-I and HLA-II. Identified peptides are derived from various canonical and non-canonical viral ORFs, are shared between different cells, and are immunogenic. As the identified peptides are unique to SARS-CoV-2, they could potentially serve as future treatment targets.
Patton E. E., Mueller K. L., Adams D. J., Anandasabapathy N., Aplin A. E., Bertolotto C., Bosenberg M., Ceol C. J., Chi P., Herlyn M., Holmen S. L., Karreth F. A., Kaufman C. K., Khan S., Kobold S., Leucci E., Levy C., Lombard D. B., Lund A. W., Marie K. L., Marine J. C., Marais R., McMahon M., Robles-Espinoza C. D., Ronai Z. A., Samuels Y., Soengas M. S., Villanueva J., Weeraratna A. T., White R. M., Yeh I., Zhu J., Zon L. I., Hurlbert M. S. & Merlino G.
(2021)
Cancer Cell.
39,
5,
p. 610-631
There is a lack of appropriate melanoma models that can be used to evaluate the efficacy of novel therapeutic modalities. Here, we discuss the current state of the art of melanoma models including genetically engineered mouse, patient-derived xenograft, zebrafish, and ex vivo and in vitro models. We also identify five major challenges that can be addressed using such models, including metastasis and tumor dormancy, drug resistance, the melanoma immune response, and the impact of aging and environmental exposures on melanoma progression and drug resistance. Additionally, we discuss the opportunity for building models for rare subtypes of melanomas, which represent an unmet critical need. Finally, we identify key recommendations for melanoma models that may improve accuracy of preclinical testing and predict efficacy in clinical trials, to help usher in the next generation of melanoma therapies.
Kalaora S., Nagler A., Nejman D., Alon M., Barbolin C., Barnea E., Ketelaars S. L., Cheng K., Vervier K., Shental N., Bussi Y., Rotkopf R., Levy R., Benedek G., Trabish S., Dadosh T., Levin-Zaidman S., Geller L. T., Wang K., Greenberg P., Yagel G., Peri A., Fuks G., Bhardwaj N., Reuben A., Hermida L., Johnson S. B., Galloway-Peña J. R., Shropshire W. C., Bernatchez C., Haymaker C., Arora R., Roitman L., Eilam R., Weinberger A., Lotan-Pompan M., Lotem M., Admon A., Levin Y., Lawley T. D., Adams D. J., Levesque M. P., Besser M. J., Schachter J., Golani O., Segal E., Geva-Zatorsky N., Ruppin E., Kvistborg P., Peterson S. N., Wargo J. A., Straussman R. & Samuels Y.
(2021)
Nature.
592,
7852,
p. 138-143
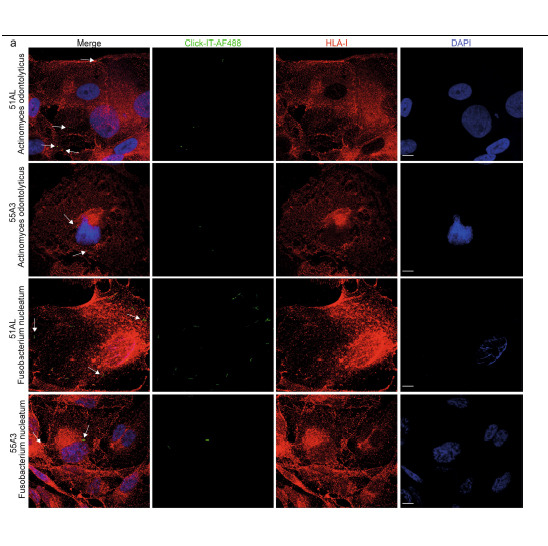
A variety of species of bacteria are known to colonize human tumours111, proliferate within them and modulate immune function, which ultimately affects the survival of patients with cancer and their responses to treatment1214. However, it is not known whether antigens derived from intracellular bacteria are presented by the human leukocyte antigen class I and II (HLA-I and HLA-II, respectively) molecules of tumour cells, or whether such antigens elicit a tumour-infiltrating T cell immune response. Here we used 16S rRNA gene sequencing and HLA peptidomics to identify a peptide repertoire derived from intracellular bacteria that was presented on HLA-I and HLA-II molecules in melanoma tumours. Our analysis of 17 melanoma metastases (derived from 9 patients) revealed 248 and 35 unique HLA-I and HLA-II peptides, respectively, that were derived from 41 species of bacteria. We identified recurrent bacterial peptides in tumours from different patients, as well as in different tumours from the same patient. Our study reveals that peptides derived from intracellular bacteria can be presented by tumour cells and elicit immune reactivity, and thus provides insight into a mechanism by which bacteria influence activation of the immune system and responses to therapy.
Yakovian O., Sajman J., Arafeh R., Neve-Oz Y., Alon M., Samuels Y. & Sherman E.
(2021)
81,
5,
p. 1279-1292
Hotspot mutations of the oncogenes BRAF and NRas are the most common genetic alterations in cutaneous melanoma. Still, the nanoscale organization and signal coupling of these proteins remain incompletely understood, particularly upon expression of oncogenic NRas mutants. Here we employed single-molecule localization microscopy to study the nanoscale organization of NRas and BRAF at the plasma membrane (PM) of melanoma cells. NRas and BRAF resided in self-clusters that did not associate well in resting cells. In EGF-activated cells, NRas clusters became more diffused while overall protein levels at the PM increased; thus allowing enhanced association of NRas and BRAF and downstream signaling. In multiple melanoma cell lines, mutant NRas resided in more pronounced self-clusters relative to wild-type (WT) NRas yet associated more with the clustered and more abundant BRAF. In cells resistant to trametinib, a clinical MEK inhibitor (MEKi), a similar coclustering of NRas and BRAF was observed upon EGF activation. Strikingly, treatment of cells expressing mutant NRas with trametinib reversed the effect of mutant NRas expression by restoring the nonoverlapping self-clusters of NRas and BRAF and by reducing their PM levels and elevated pERK levels caused by mutant NRas. Our results indicate a new mechanism for signal regulation of NRas in melanoma through its nanoscale dynamic organization and a new mechanism for MEKi function in melanoma cells carrying NRas mutations but lacking MEK mutations. Significance: Nanoscale dynamic organization of WT and mutant NRas relative to BRAF serves as a regulatory mechanism for NRas signaling and may be a viable therapeutic target for its sensitivity to MEKi.
Bartok O., Pataskar A., Nagel R., Laos M., Goldfarb E., Hayoun D., Levy R., Körner P., Kreuger I. Z. M., Champagne J., Zaal E. A., Bleijerveld O. B., Huang X., Kenski J., Wargo J., Brandis A., Levin Y., Mizrahi O., Alon M., Lebon S., Yang W., Nielsen M. M., Stern-Ginossar N., Altelaar M., Berkers C. R., Geiger T., Peeper D. S., Olweus J., Samuels Y. & Agami R.
(2021)
Nature.
590,
7845,
p. 332-337
Extensive tumour inflammation, which is reflected by high levels of infiltrating T cells and interferon-γ (IFNγ) signalling, improves the response of patients with melanoma to checkpoint immunotherapy. Many tumours, however, escape by activating cellular pathways that lead to immunosuppression. One such mechanism is the production of tryptophan metabolites along the kynurenine pathway by the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), which is induced by IFNγ. However, clinical trials using inhibition of IDO1 in combination with blockade of the PD1 pathway in patients with melanoma did not improve the efficacy of treatment compared to PD1 pathway blockade alone, pointing to an incomplete understanding of the role of IDO1 and the consequent degradation of tryptophan in mRNA translation and cancer progression. Here we used ribosome profiling in melanoma cells to investigate the effects of prolonged IFNγ treatment on mRNA translation. Notably, we observed accumulations of ribosomes downstream of tryptophan codons, along with their expected stalling at the tryptophan codon. This suggested that ribosomes bypass tryptophan codons in the absence of tryptophan. A detailed examination of these tryptophan-associated accumulations of ribosomes-which we term 'W-bumps'-showed that they were characterized by ribosomal frameshifting events. Consistently, reporter assays combined with proteomic and immunopeptidomic analyses demonstrated the induction of ribosomal frameshifting, and the generation and presentation of aberrant trans-frame peptides at the cell surface after treatment with IFNγ. Priming of naive T cells from healthy donors with aberrant peptides induced peptide-specific T cells. Together, our results suggest that IDO1-mediated depletion of tryptophan, which is induced by IFNγ, has a role in the immune recognition of melanoma cells by contributing to diversification of the peptidome landscape.
Nagler A., Vredevoogd D. W., Alon M., Cheng P. F., Trabish S., Kalaora S., Arafeh R., Goldin V., Levesque M. P., Peeper D. S. & Samuels Y.
(2020)
Pigment Cell and Melanoma Research.
33,
2,
p. 334-344
NRAS mutations are the most common alterations among RAS isoforms in cutaneous melanoma, with patients harboring these aggressive tumors having a poor prognosis and low survival rate. The main line of treatment for these patients is MAPK pathway-targeted therapies, such as MEK inhibitors, but, unfortunately, the response to these inhibitors is variable due to tumor resistance. Identifying genetic modifiers involved in resistance toward MEK-targeted therapy may assist in the development of new therapeutic strategies, enhancing treatment response and patient survival. Our whole-genome CRISPR-Cas9 knockout screen identified the target Kelch domain-containing F-Box protein 42 (FBXO42) as a factor involved in NRAS-mutant melanoma-acquired resistance to the MEK1/2 inhibitor trametinib. We further show that FBXO42, an E3 ubiquitin ligase, is involved in the TAK1 signaling pathway, possibly prompting an increase in active P38. In addition, we demonstrate that combining trametinib with the TAK1 inhibitor, takinib, is a far more efficient treatment than trametinib alone in NRAS-mutant melanoma cells. Our findings thus show a new pathway involved in NRAS-mutant melanoma resistance and provide new opportunities for novel therapeutic options.
Kalaora S., Lee J. S., Barnea E., Levy R., Greenberg P., Alon M., Yagel G., Bar Eli G., Oren R., Peri A., Patkar S., Bitton L., Rosenberg S. A., Lotem M., Levin Y., Admon A., Ruppin E. & Samuels Y.
(2020)
Nature Communications.
11,
1,
896.
Predicting the outcome of immunotherapy treatment in melanoma patients is challenging. Alterations in genes involved in antigen presentation and the interferon gamma (IFNγ) pathway play an important role in the immune response to tumors. We describe here that the overexpression of PSMB8 and PSMB9, two major components of the immunoproteasome, is predictive of better survival and improved response to immune-checkpoint inhibitors of melanoma patients. We study the mechanism underlying this connection by analyzing the antigenic peptide repertoire of cells that overexpress these subunits using HLA peptidomics. We find a higher response of patient-matched tumor infiltrating lymphocytes against antigens diferentially presented after immunoproteasome overexpression. Importantly, we find that PSMB8 and PSMB9 expression levels are much stronger predictors of melanoma patientsʼ immune response to checkpoint inhibitors than the tumors mutational burden. These results suggest that PSMB8 and PSMB9 expression levels can serve as important biomarkers for stratifying melanoma patients for immune-checkpoint treatment.
Azer E. S., Mehrabadi F. R., Malikic S., Li X. C., Bartok O., Litchfield K., Levy R., Samuels Y., Schäffer A. A., Gertz E. M., Day C. P., Pérez-Guijarro E., Marie K., Lee M. P., Merlino G., Ergun F. & Cenk Sahinalp S.
(2020)
Bioinformatics.
36,
p. I169-I176
Motivation: Recent advances in single-cell sequencing (SCS) offer an unprecedented insight into tumor emergence and evolution. Principled approaches to tumor phylogeny reconstruction via SCS data are typically based on general computational methods for solving an integer linear program, or a constraint satisfaction program, which, although guaranteeing convergence to the most likely solution, are very slow. Others based on Monte Carlo Markov Chain or alternative heuristics not only offer no such guarantee, but also are not faster in practice. As a result, novel methods that can scale up to handle the size and noise characteristics of emerging SCS data are highly desirable to fully utilize this technology. Results: We introduce PhISCS-BnB (phylogeny inference using SCS via branch and bound), a branch and bound algorithm to compute the most likely perfect phylogeny on an input genotype matrix extracted from an SCS dataset. PhISCS-BnB not only offers an optimality guarantee, but is also 10-100 times faster than the best available methods on simulated tumor SCS data. We also applied PhISCS-BnB on a recently published large melanoma dataset derived from the sublineages of a cell line involving 20 clones with 2367 mutations, which returned the optimal tumor phylogeny in
Almost thirty years ago, PI3K was discovered as a lipid kinase associated with certain oncoproteins. The first decade of research on PI3K saw the identification, purification and cloning of PI3Kα. The second decade of research was noted for the identification of some of PI3K's activators and effectors. This was accompanied by the discovery that PI3K acts as a retroviral oncogene. The third decade was known for the establishment of the direct involvement of PI3K in cancer, demonstrated by the identification of cancer-specific mutations. Efforts to target PI3K were on the rise from that moment on, accompanied by the first clinical trials for PI3K inhibitor therapies. In the fourth decade of research, PI3K-based cancer drugs will continue to emerge, as will new knowledge regarding other uncovered functions of this protein and pathway.
Wolf Y., Bartok O., Patkar S., Eli G. B., Cohen S., Litchfield K., Levy R., Jiménez-Sánchez A., Trabish S., Lee J. S., Karathia H., Barnea E., Day C., Cinnamon E., Stein I., Solomon A., Bitton L., Pérez-Guijarro E., Dubovik T., Shen-Orr S. S., Miller M. L., Merlino G., Levin Y., Pikarsky E., Eisenbach L., Admon A., Swanton C., Ruppin E. & Samuels Y.
(2019)
Cell.
179,
1,
p. 219-235
Although clonal neo-antigen burden is associated with improved response to immune therapy, the functional basis for this remains unclear. Here we study this question in a novel controlled mouse melanoma model that enables us to explore the effects of intra-tumor heterogeneity (ITH) on tumor aggressiveness and immunity independent of tumor mutational burden. Induction of UVB-derived mutations yields highly aggressive tumors with decreased anti-tumor activity. However, single-cell-derived tumors with reduced ITH are swiftly rejected. Their rejection is accompanied by increased T cell reactivity and a less suppressive microenvironment. Using phylogenetic analyses and mixing experiments of single-cell clones, we dissect two characteristics of ITH: the number of clones forming the tumor and their clonal diversity. Our analysis of melanoma patient tumor data recapitulates our results in terms of overall survival and response to immune checkpoint therapy. These findings highlight the importance of clonal mutations in robust immune surveillance and the need to quantify patient ITH to determine the response to checkpoint blockade.
Sana G., Madigan J. P., Gartner J. J., Fourrez M., Lin J., Qutob N., Narayan J., Shukla S., Ambudkar S. V., Xia D., Rosenberg S. A., Gottesman M. M., Samuels Y. & Gillet J.
(2019)
Journal of Investigative Dermatology.
139,
9,
p. 1985-1992
ABCB5 is an ABC transporter that was shown to confer low-level multidrug resistance in cancer. In this study, we show that ABCB5 was mutated in 13.75% of the 640 melanoma samples analyzed. Besides nonsense mutations, two mutation hotspots were found in the ABCB5 protein, in the drug-binding pocket and the nucleotide-binding domains. Four mutations, which are representative of the mutation pattern, were selected. ATPase assays showed that these mutations resulted in a decrease in basal ATP hydrolysis by ABCB5. To select informative melanoma cell lines, mutational profiles of the clinical samples were further analyzed. This study showed mutations in the tumor suppressor CDKN2A gene and the NRAS oncogene in 62.5% and 75%, respectively of the samples that had mutations in the ABCB5 gene. No mutation was found in the tumor suppressor PTEN gene, whereas the activating V600E mutation in the BRAF oncogene was found in 25% of the samples with a mutated ABCB5 gene. Studies in four melanoma cell lines with various genetic backgrounds showed an increase in the proliferation and migration capacity of mutant ABCB5-expressing cells, suggesting that ABCB5 plays a role in the development of melanoma as a tumor suppressor gene.
Qutob N., Masuho I., Alon M., Emmanuel R., Cohen I., Di Pizio A., Madore J., Elkahloun A., Ziv T., Levy R., Gartner J. J., Hill V. K., Lin J. C., Hevroni Y., Greenberg P., Brodezki A., Rosenberg S. A., Kosloff M., Hayward N. K., Admon A., Niv M. Y., Scolyer R. A., Martemyanov K. A. & Samuels Y.
(2019)
Scientific Reports.
9,
1,
4523.
The Acknowledgements section in this Article is incomplete.\u201cWe thank Mr. N. K. Skamangas for technical support and Dr. A. Kovoor for the D2 receptor expression plasmid, Dr. H. Itoh for GαoA and Dr. N. Lambert for sharing the Gβγ-Venus constructs. This work was supported by NIH grants DA036596 and DA026405 (KAM). MYN is supported by Israel Science Foundation grant 432/12 and Chief Scientist Ministry of Health (the ERA-NET network) and grant no. 3-9543 from the Chief Scientist Office of the Ministry of Health, Israel via the ERA-net network. MK is supported by Israel Science Foundation grant numbers 1454/13, 1959/13, 2155/15. YS is supported by the Israel Science Foundation grant numbers 1604/13 and 877/13, the ERC (StG-335377), by the Henry Chanoch Krenter Institute for Biomedical Imaging and Genomics, the estate of Alice Schwarz-Gardos, the estate of John Hunter, the Knell Family, the Peter and Patricia Gruber Award and the Hamburger Family. Lady Davis Fellowship to ADP is gratefully acknowledged. NKH and RAS are supported by fellowships from the National Health and Medical Research Council of Australia.\u201d
Arafeh R., Di Pizio A., Elkahloun A. G., Dym O., Niv M. Y. & Samuels Y.
(2019)
Oncogene.
38,
13,
p. 2432-2434
RASA2 has previously been shown to be a functional RasGAP in melanoma cells [1]. Mutation or loss of RASA2 promotes RAS activation in melanoma [1]. Our genetic analysis of RASA2 mutations identified that RASA2 and NRAS mutations are mutually exclusive (p=0.002, Fishers exact test), and that NF1 mutations [2, 3] significantly co-occur with RASA2 mutations (p=0.000011, Fishers exact test) in BRAF and NRAS wild-type melanomas, suggesting that loss of RASA2 and NF1 have complementary pro-tumorigenic functions (Fig. 1A).
Arafeh R., Flores K., Keren-Paz A., Maik-Rachline G., Gutkind N., Rosenberg S., Seger R. & Samuels Y.
(2019)
Scientific Reports.
9,
1,
4672.
Correction to the Acknowledgements section of Scientific Reports https://doi.org/10.1038/s41598-017-16558-0, published online 27 November 2017. The error has not been fixed in the paper.
Kalaora S. & Samuels Y.
(2019)
Cancer Immunosurveillance
: Methods and Protocols
.
Lopez-Soto A. & Folgueras A. R.(eds.).
Vol. 1884.
p. 203-214
(trueMethods in Molecular Biology).
Neo-antigens expressed on tumors are targets for development of cancer immunotherapy strategies. Use of prediction algorithms to identify neo-antigens yields a significant number of peptides that must be validated in laborious and time-consuming methods; many prove to be false-positive identifications. The use of HLA peptidomics allows the isolation of the HLA-peptide complexes directly from cells and can be done on fresh tumor, patient-derived xerographs, or cell lines when the tissue sample is limited. This method can be used to identify both HLA class I and HLA class II or any different MHC from different species. Here we describe the steps to create the immune-affinity columns used from the process, the immunoprecipitation procedure, and also the isolation of the peptides that will be analyzed by mass spectrometry.
The invasive phenotype of metastatic cancer cells is accompanied by the formation of actin-rich invadopodia,which adhere to the extracellular matrix and degrade it. In this study, we explored the role of the tyrosine kinome in the formation of invadopodia in metastatic melanoma cells. Using a microscopy-based siRNA screen, we identified a series of regulators, the knockdown of which either suppresses (e.g., TYK2, IGFR1, ERBB3, TYRO3, FES, ALK, PTK7) or enhances (e.g., ABL2, AXL, CSK) invadopodia formation and function. Notably, the receptor tyrosine kinase AXL displayed a dual regulatory function, where both depletion or overexpression enhanced invadopodia formation and activity. This apparent contradictionwas attributed to the capacity of AXL to directly stimulate invadopodia, yet its suppression upregulates the ERBB3 signaling pathway, which can also activate core invadopodia regulators and enhance invadopodia function. Bioinformatic analysis of multiple melanoma cell lines points to an inverse expression pattern of AXL and ERBB3. High expression of AXL in melanoma cells is associated with high expression of invadopodia components and an invasive phenotype. These results provide new insights into the complexity of metastasis-promoting mechanisms and suggest that targeting of multiple invadopodia signaling networks may serve as a potential anti-invasion therapy in melanoma. Significance: These findings uncover a unique interplay between AXL and ERBB3 in invadopodia regulation that points to the need for combined therapy in order to prevent invadopodia-mediated metastasis in melanoma.
Martin D., Degese M. S., Vitale-Cross L., Iglesias-Bartolome R., Valera J. L. C., Wang Z., Feng X., Yeerna H., Vadmal V., Moroishi T., Thorne R. F., Zaida M., Siegele B., Cheong S. C., Molinolo A. A., Samuels Y., Tamayo P., Guan K. L., Lippman S. M., Lyons J. G. & Gutkind J. S.
(2018)
Nature Communications.
9,
1,
2372.
Dysregulation of the Hippo signaling pathway and the consequent YAP1 activation is a frequent event in human malignancies, yet the underlying molecular mechanisms are still poorly understood. A pancancer analysis of core Hippo kinases and their candidate regulating molecules revealed few alterations in the canonical Hippo pathway, but very frequent genetic alterations in the FAT family of atypical cadherins. By focusing on head and neck squamous cell carcinoma (HNSCC), which displays frequent FAT1 alterations (29.8%), we provide evidence that FAT1 functional loss results in YAP1 activation. Mechanistically, we found that FAT1 assembles a multimeric Hippo signaling complex (signalome), resulting in activation of core Hippo kinases by TAOKs and consequent YAP1 inactivation. We also show that unrestrained YAP1 acts as an oncogenic driver in HNSCC, and that targeting YAP1 may represent an attractive precision therapeutic option for cancers harboring genomic alterations in the FAT1 tumor suppressor genes.
Wolf Y. & Samuels Y.
(2018)
ESMO Open.
3,
7,
e000475.
The most meaningful advancement in cancer treatment in recent years has been the emergence of immunotherapy. Checkpoint inhibitor blockade and adoptive T cell therapy have shown remarkable clinical effects in a wide range of tumour types. Despite these advances, many tumours do not respond to these treatments, which raises the need to further investigate how patients can benefit from immunotherapy. This effort can now take advantage of the recent technological progress in single-cell, high-throughput sequencing and computational efforts. In this review, we will discuss advances in different immunotherapies and the principles of cancer immunogenomics, with an emphasis on the detection of cancer neoantigens with human leucocyte antigen peptidomics, and how these principles can be further used for more efficient clinical output.
Kalaora S., Wolf Y., Feferman T., Barnea E., Greenstein E., Reshef D., Tirosh I., Reuben A., Patkar S., Levy R., Quinkhardt J., Omokoko T., Qutob N., Golani O., Zhang J., Mao X., Song X., Bernatchez C., Haymaker C., Forget M., Creasy C., Greenberg P., Carter B. W., Cooper Z. A., Rosenberg S. A., Lotem M., Sahin U., Shakhar G., Ruppin E., Wargo J. A., Friedman N., Admon A. & Samuels Y.
(2018)
Cancer Discovery.
8,
11,
p. 1366-1375
The quest for tumor-associated antigens (TAA) and neoantigens is a major focus of cancer immunotherapy. Here, we combine a neoantigen prediction pipeline and human leukocyte antigen (HLA) peptidomics to identify TAAs and neoantigens in 16 tumors derived from seven patients with melanoma and characterize their interactions with their tumor-infiltrating lymphocytes (TIL). Our investigation of the antigenic and T-cell landscapes encompassing the TAA and neoantigen signatures, their immune reactivity, and their corresponding T-cell identities provides the first comprehensive analysis of cancer cell T-cell cosignatures, allowing us to discover remarkable antigenic and TIL similarities between metastases from the same patient. Furthermore, we reveal that two neoantigen-specific clonotypes killed 90% of autologous melanoma cells, both in vitro and in vivo, showing that a limited set of neoantigen-specific T cells may play a central role in melanoma tumor rejection. Our findings indicate that combining HLA peptidomics with neoantigen predictions allows robust identification of targetable neoantigens, which could successfully guide personalized cancer immunotherapies.SIGNIFICANCE: As neoantigen targeting is becoming more established as a powerful therapeutic approach, investigating these molecules has taken center stage. Here, we show that a limited set of neoantigen-specific T cells mediates tumor rejection, suggesting that identifying just a few antigens and their corresponding T-cell clones could guide personalized immunotherapy. (c) 2018 AACR.
Nordlinger A., Dror S., Elkahloun A., Del Rio J., Stubbs E., Golan T., Malcov H., Pricket T. D., Cronin J. C., Parikh S., Labes S., Thomas L., Yankovitz G., Tabach Y., Levy C., Samuels Y. & Khaled M.
(2018)
Journal of Investigative Dermatology.
138,
10,
p. 2216-2223
Melanoma, a melanocyte origin neoplasm, is the most lethal type of skin cancer, and incidence is increasing. Several familial and somatic mutations have been identified in the gene encoding the melanocyte lineage master regulator, MITF; however, the neoplastic mechanisms of these mutant MITF variants are mostly unknown. Here, by performing unbiased analysis of the transcriptomes in cells expressing mutant MITF, we identified calcium-binding protein S100A4 as a downstream target of MITF-E87R. By using wild-type and mutant MITF melanoma lines, we found that both endogenous wild-type and MITF-E87R variants occupy the S100A4 promoter. Remarkably, whereas wild-type MITF represses S100A4 expression, MITF-E87R activates its transcription. The opposite effects of wild-type and mutant MITF result in opposing cellular phenotypes, because MITF-E87R via S100A4 enhanced invasion and reduced adhesion in contrast to wild-type MITF activity. Finally, we found that melanoma patients with altered S100A4 expression have poor prognosis. These data show that a change in MITF transcriptional activity from repression to activation of S100A4 that results from a point mutation in MITF alters melanoma invasive ability. These data suggest new opportunities for diagnosis and treatment of metastatic melanoma.
Lee J. S., Adler L., Karathia H., Carmel N., Rabinovich S., Auslander N., Keshet R., Stettner N., Silberman A., Agemy L., Helbling D., Eilam R., Sun Q., Brandis A., Malitsky S., Itkin M., Weiss H., Pinto S., Kalaora S., Levy R., Barnea E., Admon A., Dimmock D., Stern-Ginossar N., Scherz A., Nagamani S. C. S., Unda M., Wilson D. M., Elhasid R., Carracedo A., Samuels Y., Hannenhalli S., Ruppin E. & Erez A.
(2018)
Cell.
174,
6,
p. 1559-1570
The urea cycle (UC) is the main pathway by which mammals dispose of waste nitrogen. We find that specific alterations in the expression of most UC enzymes occur in many tumors, leading to a general metabolic hallmark termed \u201cUC dysregulation\u201d (UCD). UCD elicits nitrogen diversion toward carbamoyl-phosphate synthetase2, aspartate transcarbamylase, and dihydrooratase (CAD) activation and enhances pyrimidine synthesis, resulting in detectable changes in nitrogen metabolites in both patient tumors and their bio-fluids. The accompanying excess of pyrimidine versus purine nucleotides results in a genomic signature consisting of transversion mutations at the DNA, RNA, and protein levels. This mutational bias is associated with increased numbers of hydrophobic tumor antigens and a better response to immune checkpoint inhibitors independent of mutational load. Taken together, our findings demonstrate that UCD is a common feature of tumors that profoundly affects carcinogenesis, mutagenesis, and immunotherapy response. Urea cycle dysregulation (UCD) in cancer is a prevalent phenomenon in multiple cancers. UCD increases nitrogen utilization for pyrimidine synthesis, generating nucleotide imbalance that leads to detectable mutation patterns and biochemical signatures in cancer patients samples. UCD is associated with a worse prognosis but a better response to immunotherapy.
Alon M., Emmanuel R., Qutob N., Bakhman A., Peshti V., Brodezki A., Bassan D., Kosloff M. & Samuels Y.
(2018)
Pigment Cell and Melanoma Research.
31,
5,
p. 641-648
The NRAS oncoprotein is highly mutated in melanoma. However, to date, no comprehensive proteomic study has been reported for NRAS. Here, we utilized the endogenous epitope tagging (EET) approach for the identification of novel NRAS binding partners. Using EET, an epitope tag is added to the endogenously expressed protein, via modification of its genomic coding sequence. Existing EET systems are not robust, suffer from high background, and are labor-intensive. To this end, we present a polyadenylation signal-trap construct for N'-tagging that generates a polycistronic mRNA with the gene of interest. This system requires the integration of the tagging cassette in frame with the target gene to be expressed. Using this design, we demonstrate, for the first time, endogenous tagging of NRAS in melanoma cells allowing the identification of the E3 ubiquitin ligase c-CBL as a novel NRAS binding partner. Thus, our developed EET technology allows the characterization of new RAS effectors, which could be beneficial for the design of future drugs that inhibit constitutive signaling of RAS oncogenic mutants.
Alon M., Arafeh R., Lee J. S., Madan S., Kalaora S., Nagler A., Abgarian T., Greenberg P., Ruppin E. & Samuels Y.
(2018)
Oncotarget.
9,
58,
p. 31264-31277
Neurofibromin 1 (NF1), a tumor suppressor that negatively regulates RAS through its GTPase activity, is highly mutated in various types of sporadic human cancers, including melanoma. However, the binding partners of NF1 and the pathways in which it is involved in melanoma have not been characterized in an in depth manner. Utilizing a mass spectrometry analysis of NF1 binding partners, we revealed Calpain1 (CAPN1), a calcium-dependent neutral cysteine protease, as a novel NF1 binding partner that regulates NF1 degradation in melanoma cells. ShRNA-mediated knockdown of CAPN1 or treatment with a CAPN1 inhibitor stabilizes NF1 protein levels, downregulates AKT signaling and melanoma cell growth. Combination treatment of Calpain inhibitor I with MEKi Trametinib in different melanoma cells is more effective in reducing melanoma cell growth compared to treatment with Trametinib alone, suggesting that this combination may have a therapeutic potential in melanoma. This novel mechanism for regulating NF1 in melanoma provides a molecular basis for targeting CAPN1 in order to stabilize NF1 levels and, in doing so, suppressing Ras activation; this mechanism can be exploited therapeutically in melanoma and other cancers.
Lee J. S., Das A., Jerby-Arnon L., Arafeh R., Auslander N., Davidson M., McGarry L., James D., Amzallag A., Park S. G., Cheng K., Robinson W., Atias D., Stossel C., Buzhor E., Stein G., Waterfall J. J., Meltzer P. S., Golan T., Hannenhalli S., Gottlieb E., Benes C. H., Samuels Y., Shanks E. & Ruppin E.
(2018)
Nature Communications.
9,
1,
2546.
While synthetic lethality (SL) holds promise in developing effective cancer therapies, SL candidates found via experimental screens often have limited translational value. Here we present a data-driven approach, ISLE (identification of clinically relevant synthetic lethality), that mines TCGA cohort to identify the most likely clinically relevant SL interactions (cSLi) from a given candidate set of lab-screened SLi. We first validate ISLE via a benchmark of large-scale drug response screens and by predicting drug efficacy in mouse xenograft models. We then experimentally test a select set of predicted cSLi via new screening experiments, validating their predicted context-specific sensitivity in hypoxic vs normoxic conditions and demonstrating cSLi's utility in predicting synergistic drug combinations. We show that cSLi can successfully predict patients' drug treatment response and provide patient stratification signatures. ISLE thus complements existing actionable mutation-based methods for precision cancer therapy, offering an opportunity to expand its scope to the whole genome.
Yu Y., Schleich K., Yue B., Ji S., Lohneis P., Kemper K., Silvis M. R., Qutob N., van Rooijen E., Werner-Klein M., Li L., Dhawan D., Meierjohann S., Reimann M., Elkahloun A., Treitschke S., Dörken B., Speck C., Mallette F. A., Zon L. I., Holmen S. L., Peeper D. S., Samuels Y., Schmitt C. A. & Lee S.
(2018)
Cancer Cell.
33,
2,
p. 322-336.e8
Oncogene-induced senescence, e.g., in melanocytic nevi, terminates the expansion of pre-malignant cells via transcriptional silencing of proliferation-related genes due to decoration of their promoters with repressive trimethylated histone H3 lysine 9 (H3K9) marks. We show here that structurally distinct H3K9-active demethylasesthe lysine-specific demethylase-1 (LSD1) and several Jumonji C domain-containing moieties (such as JMJD2C)disable senescence and permit Ras/Braf-evoked transformation. In mouse and zebrafish models, enforced LSD1 or JMJD2C expression promoted Braf-V600E-driven melanomagenesis. A large subset of established melanoma cell lines and primary human melanoma samples presented with a collective upregulation of related and unrelated H3K9 demethylase activities, whose targeted inhibition restored senescence, even in Braf inhibitor-resistant melanomas, evoked secondary immune effects and controlled tumor growth in vivo. Yu et al. show that two different types of histone H3 lysine 9 (H3K9) demethylases, LSD1 and JMJD2C, disable oncogenic Ras- or Braf-induced senescence by enabling the expression of E2F target genes, which permits transformation. Inhibition of the H3K9 demethylases restores senescence and controls tumor growth.
Qutob N., Masuho I., Alon M., Emmanuel R., Cohen I., Di Pizio A., Madore J., Elkahloun A., Ziv T., Levy R., Gartner J. J., Hill V. K., Lin J. C., Hevroni Y., Greenberg P., Brodezki A., Rosenberg S. A., Kosloff M., Hayward N. K., Admon A., Niv M. Y., Scolyer R. A., Martemyanov K. A. & Samuels Y.
(2018)
Scientific Reports.
8,
1,
653.
Analysis of 501 melanoma exomes revealed RGS7, which encodes a GTPase-accelerating protein (GAP), to be a tumor-suppressor gene. RGS7 was mutated in 11% of melanomas and was found to harbor three recurrent mutations (p.R44C, p.E383K and p.R416Q). Structural modeling of the most common recurrent mutation of the three (p.R44C) predicted that it destabilizes the protein due to the loss of an H-bond and salt bridge network between the mutated position and the serine and aspartic acid residues at positions 58 as 61, respectively. We experimentally confirmed this prediction showing that the p.R44C mutant protein is indeed destabilized. We further show RGS7 p.R44C has weaker catalytic activity for its substrate G alpha(o), thus providing a dual mechanism for its loss of function. Both of these effects are expected to contribute to loss of function of RGS7 resulting in increased anchorage-independent growth, migration and invasion of melanoma cells. By mutating position 56 in the R44C mutant from valine to cysteine, thereby enabling the formation of a disulfide bridge between the two mutated positions, we slightly increased the catalytic activity and reinstated protein stability, leading to the rescue of RGS7's function as a tumor suppressor. Our findings identify RGS7 as a novel melanoma driver and point to the clinical relevance of using strategies to stabilize the protein and, thereby, restore its function.
Walia V. & Samuels Y.
(2018)
Proteases and Cancer
.
Obaya A. & Cal S.(eds.).
p. 97-106
(trueMethods in Molecular Biology).
Protein zymography is the most commonly used technique to study the enzymatic activity of matrix metalloproteinases (MMPs) and their inhibitors. MMPs are proteolytic enzymes that promote extracellular matrix degradation. MMPs are frequently mutated in malignant melanomas as well as other cancers and are linked to increasing incidence of tumor metastasis. Substrate zymography characterizes MMP activity by their ability to degrade preferred substrates. Here we describe the collagen zymography technique to measure the active or latent form of MMPs using MMP-8 as an example, which is a frequently mutated MMP family member in malignant melanomas. The same technique can be used with the modification of substrate to detect metalloproteinase activity of other MMPs. Both wild-type and mutated forms of MMPs can be analyzed using a single gel using this method.
Arafeh R., Flores K., Keren-Paz A., Maik-Rachline G., Gutkind N., Rosenberg S., Seger R. & Samuels Y.
(2017)
Scientific Reports.
7,
1,
16345.
Genetic alterations in BRAF, NRAS and NF1 that activate the ERK cascade, account for over 80% of metastatic melanomas. However, ERK cascade inhibitors have been proven beneficial almost exclusively for BRAF mutant melanomas. One of the hallmarks of the ERK cascade is the nuclear translocation of ERK1/2, which is important mainly for the induction of proliferation. This translocation can be inhibited by the NTS-derived peptide (EPE) that blocks the ERK1/2-importin7 interaction, inhibits the nuclear translocation of ERK1/2, and arrests active ERK1/2 in the cytoplasm. In this study, we found that the EPE peptide significantly reduced the viability of not only BRAF, but also several NRAS and NF1 mutant melanomas. Importantly, combination of the EPE peptide and trametinib showed synergy in reducing the viability of some NRAS mutant melanomas, an effect driven by the partial preservation of negative feedback loops. The same combination significantly reduced the viability of other melanoma cells, including those resistant to mono-treatment with EPE peptide and ERK cascade inhibitors. Our study indicates that targeting the nuclear translocation of ERK1/2, in combination with MEK inhibitors can be used for the treatment of different mutant melanomas.
Prickett T. D., Gartner J. J. & Samuels Y.
(2017)
NMDA Receptors
.
p. 93-116
Ionotropic glutamate receptors (iGluRs) are large integral membrane multi-protein complexes that create ion channels in plasma membranes. Upon binding of receptor specific ligands (e.g., glutamate), increased efflux or influx of mono- or divalent cations (e.g., Ca2+) promotes synaptic transmission, cellular migration, and survival. Three classes of iGluRs were originally defined after their respective agonists: AMPA, kainate, and NMDA receptors (NMDARs). Recently, we examined iGluR families at the genetic level using Next-Generation Sequencing (NGS) (whole-exome sequencing (WES)) and discovered a high prevalence of somatic mutations within the gene for one of the NMDAR subunits, GRIN2A, specifically in malignant melanoma. Following confirmation of the somatic mutations, we focused on functional characterization of a subset of the GRIN2A mutants that demonstrated a loss of NMDAR functionality. We used gene expression and protein biochemistry to examine complex formation between GluN1 subunit (encoded by GRIN1) and GluN2A subunit (encoded by GRIN2A), anchorage-independent growth in soft agar and cellular migration. Furthermore, we used shRNA depletion of endogenous GRIN2A in melanoma cells expressing either wild-type GRIN2A or mutant GRIN2A and measured cellular proliferation compared to negative controls. Our data show that somatic mutation of certain residues in GluN2A results in increased survival and is the first such report to demonstrate the functional importance of GRIN2A mutations in melanoma and the significance ionotropic glutamate receptor signaling plays in malignant melanoma.
Soussi T., Taschner P. E. M. & Samuels Y.
(2017)
Human Mutation.
38,
4,
p. 339-342
Single-nucleotide variants (SNVs) are the most frequent genetic changes found in human cancer. Most driver alterations are missense and nonsense variants localized in the coding region of cancer genes. Unbiased cancer genome sequencing shows that synonymous SNVs (sSNVs) can be found clustered in the coding regions of several cancer oncogenes or tumor suppressor genes suggesting purifying selection. sSNVs are currently underestimated, as they are usually discarded during analysis. Furthermore, several public databases do not display sSNVs, which can lead to analytical bias and the false assumption that this mutational event is uncommon. Recent progress in our understanding of the deleterious consequences of these sSNVs for RNA stability and protein translation shows that they can act as strong drivers of cancer, as demonstrated for several cancer genes such as TP53 or BCL2L12. It is therefore essential that sSNVs be properly reported and analyzed in order to provide an accurate picture of the genetic landscape of the cancer genome. (C) 2016 Wiley Periodicals, Inc.
Samuels Y. & Emmanuel R.
(2016)
IPC No. G01N 33/ 53 A I,
Patent No. WO2016113733,
17 Nov 2015,
Priority No. US201562256153P
There are provided tagging vectors, compositions comprising the same and methods of using the same for specific and efficient endogenous epitope tagging of target genes in target cells.
Levin L., Srour S., Gartner J., Kapitansky O., Qutob N., Dror S., Golan T., Dayan R., Brener R., Ziv T., Khaled M., Schueler-Furman O., Samuels Y. & Levy C.
(2016)
Journal of Genetics and Genomics.
43,
6,
p. 369-379
Epidemiological studies suggest a direct link between melanoma and Parkinson's disease (PD); however, the underlying molecular basis is unknown. Since mutations in Parkin are the major driver of early-onset PD and Parkin was recently reported to play a role in cancer development, we hypothesized that Parkin links melanoma and PD. By analyzing whole exome/genome sequencing of Parkin from 246 melanoma patients, we identified five non-synonymous mutations, three synonymous mutations, and one splice region variant in Parkin in 3.6% of the samples. In vitro analysis showed that wild-type Parkin plays a tumor suppressive role in melanoma development resulting in cell-cycle arrest, reduction of metabolic activity, and apoptosis. Using a mass spectrometry-based analysis, we identified potential Parkin substrates in melanoma and generated a functional protein association network. The activity of mutated Parkin was assessed by protein structure modeling and examination of Parkin E3 ligase activity. The Parkin-E28K mutation impairs Parkin ubiquitination activity and abolishes its tumor suppressive effect. Taken together, our analysis of genomic sequence and in vitro data indicate that Parkin is a potential link between melanoma and Parkinson's disease. Our findings suggest new approaches for early diagnosis and treatment against both diseases.
Hao Y., Samuels Y., Li Q., Krokowski D., Guan B., Wang C., Jin Z., Dong B., Cao B., Feng X., Xiang M., Xu C., Fink S., Meropol N. J., Xu Y., Conlon R. A., Markowitz S., Kinzler K. W., Velculescu V. E., Brunengraber H., Willis J. E., LaFramboise T., Hatzoglou M., Zhang G., Vogelstein B. & Wang Z.
(2016)
Nature Communications.
7,
11971.
Cancer cells often require glutamine for growth, thereby distinguishing them from most normal cells. Here we show that PIK3CA mutations reprogram glutamine metabolism by upregulating glutamate pyruvate transaminase 2 (GPT2) in colorectal cancer (CRC) cells, making them more dependent on glutamine. Compared with isogenic wild-type (WT) cells, PIK3CA mutant CRCs convert substantially more glutamine to α-ketoglutarate to replenish the tricarboxylic acid cycle and generate ATP. Mutant p110α upregulates GPT2 gene expression through an AKT-independent, PDK1-RSK2-ATF4 signalling axis. Moreover, aminooxyacetate, which inhibits the enzymatic activity of aminotransferases including GPT2, suppresses xenograft tumour growth of CRCs with PIK3CA mutations, but not with WT PIK3CA. Together, these data establish oncogenic PIK3CA mutations as a cause of glutamine dependency in CRCs and suggest that targeting glutamine metabolism may be an effective approach to treat CRC patients harbouring PIK3CA mutations.
Inzelberg R., Samuels Y., Azizi E., Qutob N., Inzelberg L., Domany E., Schechtman E. & Friedman E.
(2016)
Neurology: Genetics.
2,
3,
e70.
Objective: To assess whether Parkinson disease (PD) genes are somatically mutated in cutaneous melanoma (CM) tissue, because CM occurs in patients with PD at higher rates than in the general population and PD is more common than expected in CM cohorts. Methods: We cross-referenced somatic mutations in metastatic CM detected by whole-exome sequencing with the 15 known PD (PARK) genes. We computed the empirical distribution of the sum of mutations in each gene (Smut) and of the number of tissue samples in which a given gene was mutated at least once (SSampl) for each of the analyzable genes, determined the 90th and 95th percentiles of the empirical distributions of these sums, and verified the location of PARK genes in these distributions. Identical analyses were applied to adenocarcinoma of lung (ADENOCA-LUNG) and squamous cell carcinoma of lung (SQUAMCA-LUNG). We also analyzed the distribution of the number of mutated PARK genes in CM samples vs the 2 lung cancers. Results: Somatic CM mutation analysis (n 246) detected 315,914 mutations in 18,758 genes. Somatic CM mutations were found in 14 of 15 PARK genes. Forty-eight percent of CM samples carried ≥1 PARK mutation and 25% carried multiple PARK mutations. PARK8 mutations occurred above the 95th percentile of the empirical distribution for SMut and SSampl. Significantly more CM samples harbored multiple PARK gene mutations compared with SQUAMCA-LUNG (p 0.0026) and with ADENOCA-LUNG (p < 0.0001). Conclusions: The overrepresentation of somatic PARK mutations in CM suggests shared dysregulated pathways for CM and PD.
Revach O. Y., Winograd-Katz S. E., Samuels Y. & Geiger B.
(2016)
Experimental Cell Research.
343,
1,
p. 82-88
In this article, we discuss the complex involvement of a Rho-family GTPase, Rac1, in cell migration and in invadopodia-mediated matrix degradation. We discuss the involvement of invadopodia in invasive cell migration, and their capacity to promote cancer metastasis. Considering the regulation of invadopodia formation, we describe studies that demonstrate the role of Rac1 in the metastatic process, and the suggestion that this effect is attributable to the capacity of Rac1 to promote invadopodia formation. This notion is demonstrated here by showing that knockdown of Rac1 in melanoma cells expressing a wild-type form of this GTPase, reduces invadopodia-dependent matrix degradation. Interestingly, we also show that excessive activity of Rac1, displayed by the P29S, hyperactive, "fast cycling" mutant of Rac1, which is present in 5-10% of melanoma tumors, inhibits invadopodia function. Moreover, knockdown of this hyperactive mutant enhanced matrix degradation, indicating that excessive Rac1 activity by this mutant can negatively regulate invadopodia formation and function.
The Cancer Genome
Samuels Y., Bardelli A., Gartner J. J. & López-Otin C.
(2016)
Cancer of the Breast
: Cancer: Principles and Practice of Oncology, 10th Edition
.
p. 2-23
The Cancer Genome
Samuels Y., Bardelli A., Gartner J. J. & López-Otin C.
(2016)
Colon and Other Gastrointestinal Cancers
: Cancer: Principles and Practice of Oncology, 10th edition
.
p. 2-23
Kalaora S., Barnea E., Merhavi-Shoham E., Qutob N., Teer J. K., Shimony N., Schachter J., Rosenberg S. A., Besser M. J., Admon A. & Samuels Y.
(2016)
Oncotarget.
7,
5,
p. 5110-5117
The antigenicity of cells is demarcated by the peptides bound by their Human Leucocyte Antigen (HLA) molecules. Through this antigen presentation, T cell specificity response is controlled. As a fraction of the expressed mutated peptides is presented on the HLA, these neo-epitopes could be immunogenic. Such neo-antigens have recently been identified through screening for predicted mutated peptides, using synthetic peptides or ones expressed from minigenes, combined with screening of patient tumor-infiltrating lymphocytes (TILs). Here we present a time and cost-effective method that combines whole-exome sequencing analysis with HLA peptidome mass spectrometry, to identify neo-antigens in a melanoma patient. Of the 1,019 amino acid changes identified through exome sequencing, two were confirmed by mass spectrometry to be presented by the cells. We then synthesized peptides and evaluated the two mutated neo-antigens for reactivity with autologous bulk TILs, and found that one yielded mutant-specific T-cell response. Our results demonstrate that this method can be used for immune response prediction and promise to provide an alternative approach for identifying immunogenic neo-epitopes in cancer.
Arafeh R., Qutob N., Emmanuel R., Keren-Paz A., Madore J., Elkahloun A., Wilmott J. S., Gartner J. J., Di Pizio A., Winograd-Katz S., Sindiri S., Rotkopf R., Dutton-Regester K., Johansson P., Pritchard A. L., Waddell N., Hill V. K., Lin J. C., Hevroni Y., Rosenberg S. A., Khan J., Ben-Dor S., Niv M. Y., Ulitsky I., Mann G. J., Scolyer R. A., Hayward N. K. & Samuels Y.
(2015)
Nature Genetics.
47,
12,
p. 1408-1410
Analysis of 501 melanoma exomes identified RASA2, encoding a RasGAP, as a tumor-suppressor gene mutated in 5% of melanomas. Recurrent loss-of-function mutations in RASA2 were found to increase RAS activation, melanoma cell growth and migration. RASA2 expression was lost in >= 30% of human melanomas and was associated with reduced patient survival. These findings identify RASA2 inactivation as a melanoma driver and highlight the importance of RasGAPs in cancer.
Gotea V., Gartner J. J., Qutob N., Elnitski L. & Samuels Y.
(2015)
Pigment Cell and Melanoma Research.
28,
6,
p. 673-684
Recent technological advances in sequencing have flooded the field of cancer research with knowledge about somatic mutations for many different cancer types. Most cancer genomics studies focus on mutations that alter the amino acid sequence, ignoring the potential impact of synonymous mutations. However, accumulating experimental evidence has demonstrated clear consequences for gene function, leading to a widespread recognition of the functional role of synonymous mutations and their causal connection to various diseases. Here, we review the evidence supporting the direct impact of synonymous mutations on gene function via gene splicing; mRNA stability, folding, and translation; protein folding; and miRNA-based regulation of expression. These results highlight the functional contribution of synonymous mutations to oncogenesis and the need to further investigate their detection and prioritization for experimental assessment.
Kim H., Frederick D., Levesque M., Cooper Z., Feng Y., Krepler C., Brill L., Samuels Y., Hayward N., Perlina A., Piris A., Zhang T., Halaban R., Herlyn M., Brown K., Wargo J., Dummer R., Flaherty K. & Ronai Z.
(2015)
Cell Reports.
11,
9,
p. 1458-1473
Despite the remarkable clinical response of melanoma harboring BRAF mutations to BRAF inhibitors (BRAFi), most tumors become resistant. Here, we identified the downregulation of the ubiquitin ligase RNF125 in BRAFi-resistant melanomas and demonstrated its role in intrinsic and adaptive resistance to BRAFi in cultures as well as its association with resistance in tumor specimens. Sox10/MITF expression correlated with and contributed to RNF125 transcription. Reduced RNF125 was associated with elevated expression of receptor tyrosine kinases (RTKs), including EGFR. Notably, RNF125 altered RTK expression through JAK1, which we identified as an RNF125 substrate. RNF125 bound to and ubiquitinated JAK1, prompting its degradation and suppressing RTK expression. Inhibition of JAK1 and EGFR signaling overcame BRAFi resistance in melanoma with reduced RNF125 expression, as shown in culture and in in vivo xenografts. Our findings suggest that combination therapies targeting both JAK1 and EGFR could be effective against BRAFi-resistant tumors with de novo low RNF125 expression.
Akbani R., Akdemir K. C., Aksoy B. A., Albert M., Ally A., Amin S. B., Arachchi H., Arora A., Auman J. T., Ayala B., Baboud J., Balasundaram M., Balu S., Barnabas N., Bartlett J., Bartlett P., Bastian B. C., Baylin S. B., Behera M. & Samuels Y.
(2015)
Cell.
161,
7,
p. 1681-1696
Summary We describe the landscape of genomic alterations in cutaneous melanomas through DNA, RNA, and protein-based analysis of 333 primary and/or metastatic melanomas from 331 patients. We establish a framework for genomic classification into one of four subtypes based on the pattern of the most prevalent significantly mutated genes: mutant BRAF, mutant RAS, mutant NF1, and Triple-WT (wild-type). Integrative analysis reveals enrichment of KIT mutations and focal amplifications and complex structural rearrangements as a feature of the Triple-WT subtype. We found no significant outcome correlation with genomic classification, but samples assigned a transcriptomic subclass enriched for immune gene expression associated with lymphocyte infiltrate on pathology review and high LCK protein expression, a T cell marker, were associated with improved patient survival. This clinicopathological and multi-dimensional analysis suggests that the prognosis of melanoma patients with regional metastases is influenced by tumor stroma immunobiology, offering insights to further personalize therapeutic decision-making.
Keren-Paz A., Emmanuel R. & Samuels Y.
(2015)
Nature Genetics.
47,
3,
p. 193-194
Deciphering mechanisms of drug resistance is crucial to winning the battle against cancer. A new study points to an unexpected function of YAP in drug resistance and illuminates its potential role as a therapeutic target.
The cancer genome
Samuels Y., Bardelli A., Gartner J. J. & López-Otin C.
(2015)
DeVita, Hellman, and Rosenberg's Cancer: Principles & Practice of Oncology: Tenth Edition
.
10th ed.
Walia V., Prickett T. D., Kim J. S., Gartner J. J., Lin J. C., Zhou M., Rosenberg S. A., Elble R. C., Solomon D. A., Waldman T. & Samuels Y.
(2014)
Human Mutation.
35,
11,
p. 1301-1310
Protein tyrosine phosphatases (PTPs) tightly regulate tyrosine phosphorylation essential for cell growth, adhesion, migration, and survival. We performed a mutational analysis of the PTP gene family in cutaneous metastatic melanoma and identified 23 phosphatase genes harboring somatic mutations. Among these, receptor-type tyrosine-protein phosphatase delta (PTPRD) was one of the most highly mutated genes, harboring 17 somatic mutations in 79 samples, a prevalence of 21.5%. Functional evaluation of six PTPRD mutations revealed enhanced anchorage-dependent and anchorage-independent growth. Interestingly, melanoma cells expressing mutant PTPRD were significantly more migratory than cells expressing wild-type PTPRD or vector alone, indicating a novel gain-of-function associated with mutant PTPRD. To understand the molecular mechanisms of PTPRD mutations, we searched for its binding partners by converting the active PTPRD enzyme into a "substrate trap" form. Using mass spectrometry and coimmunoprecipitation, we report desmoplakin, a desmosomal protein that is implicated in cell-cell adhesion, as a novel PTPRD substrate. Further analysis showed reduced phosphatase activity of mutant PTPRD against desmoplakin. Our findings identify an essential signaling cascade that is disrupted in melanoma. Moreover, because PTPRD is also mutated in glioblastomas and adenocarcinoma of the colon and lung, our data might be applicable to a large number of human cancers.
Prickett T. D., Zerlanko B. J., Hill V. K., Gartner J. J., Qutob N., Jiang J., Simaan M., Wunderlich J., Gutkind J. S., Rosenberg S. A. & Samuels Y.
(2014)
Journal of Investigative Dermatology.
134,
9,
p. 2390-2398
The ionotropic glutamate receptors (N-methyl-D-aspartate receptors (NMDARs)) are composed of large complexes of multi-protein subunits creating ion channels in the cell plasma membranes that allow for influx or efflux of mono- or divalent cations (e.g., Ca2+) important for synaptic transmissions, cellular migration, and survival. Recently, we discovered the high prevalence of somatic mutations within one of the ionotropic glutamate receptors, GRIN2A, in malignant melanoma. Functional characterization of a subset of GRIN2A mutants demonstrated a loss of NMDAR complex formation between GRIN1 and GRIN2A, increased anchorage-independent growth in soft agar, and increased migration. Somatic mutation of GRIN2A results in a dominant negative effect inhibiting the tumor-suppressive phenotype of wild-type (WT) GRIN2A in melanoma. Depletion of endogenous GRIN2A in melanoma cells expressing WT GRIN2A resulted in increased proliferation compared with control. In contrast, short-hairpin RNA depletion of GRIN2A in mutant cell lines slightly reduced proliferation. Our data show that somatic mutation of GRIN2A results in increased survival, and we demonstrate the functional importance of GRIN2A mutations in melanoma and the significance that ionotropic glutamate receptor signaling has in malignant melanoma.
Lu Y. C., Yao X., Crystal J. S., Li Y. F., El-Gamil M., Gross C., Davis L., Dudley M. E., Yang J. C., Samuels Y., Rosenberg S. A. & Robbins P. F.
(2014)
Clinical Cancer Research.
20,
13,
p. 3401-3410
Purpose: Cancer immunotherapy with adoptive transfer of tumor-infiltrating lymphocytes (TIL) represents an effective treatment for patients with metastatic melanoma, with the objective regressions in up to 72% of patients in three clinical trials. However, the antigen targets recognized by these effective TILs remain largely unclear. Experimental Design: Melanoma patients 2359 and 2591 both experienced durable complete regressions of metastases ongoing beyond five years following adoptive TIL transfer. Two conventional screening approaches were carried out to identify the antigens recognized by these clinically effective TILs. In addition, a novel approach was developed in this study to identify mutated T-cell antigens by screening a tandem minigene library, which comprised nonsynonymous mutation sequences identified by whole-exome sequencing of autologous tumors. Results: Screening of an autologous melanoma cDNA library using a conventional approach led to the identification of previously undescribed nonmutated targets recognized by TIL 2359 or TIL 2591. In contrast, screening of tandem minigene libraries encoding tumor-specific mutations resulted in the identification of mutated kinesin family member 2C (KIF2C) antigen as a target of TIL 2359, and mutated DNA polymerase alpha subunit B (POLA2) antigen as a target of TIL 2591. Both KIF2C and POLA2 have been found to play important roles in cell proliferation. Conclusions: These findings suggest that the minigene screening approach can facilitate the antigen repertoire analysis of tumor reactive T cells, and lead to the development of new adoptive cell therapies with purified T cells that recognize candidate-mutated antigens derived from genes essential for the carcino-genesis.
Prickett T. D., Zerlanko B., Gartner J. J., Parker S. C. J., Dutton-Regester K., Lin J. C., Teer J. K., Wei X., Jiang J., Chen G., Davies M. A., Gershenwald J. E., Robinson W., Robinson S., Hayward N. K., Rosenberg S. A., Margulies E. H. & Samuels Y.
(2014)
Journal of Investigative Dermatology.
134,
2,
p. 452-460
Patients with advanced metastatic melanoma have poor prognosis and the genetics underlying its pathogenesis are poorly understood. High-throughput sequencing has allowed comprehensive discovery of somatic mutations in cancer samples. Here, on analysis of our whole-genome and whole-exome sequencing data of 29 melanoma samples, we identified several genes that harbor recurrent nonsynonymous mutations. These included MAP3K5 (mitogen-activated protein kinase kinase kinase-5), which in a prevalence screen of 288 melanomas was found to harbor a R256C substitution in 5 cases. All MAP3K5-mutated samples were wild type for BRAF, suggesting a mutual exclusivity for these mutations. Functional analysis of the MAP3K5 R256C mutation revealed attenuation of MKK4 (mitogen-activated protein kinase kinase 4) activation through increased binding of the inhibitory protein thioredoxin (TXN/TRX-1/Trx), resulting in increased proliferation and anchorage-independent growth of melanoma cells. This mutation represents a potential target for the design of new therapies to treat melanoma.
Dutton-Regester K., Gartner J. J., Emmanuel R., Qutob N., Davies M. A., Gershenwald J. E., Robinson W., Robinson S., Steven A. R., Scolyer R. A., Mann G. J., Thompson J. F., Hayward N. K. & Samuels Y.
(2014)
Oncotarget.
5,
10,
p. 2912-2917
The incidence of melanoma continues to rise globally and is increasing at a rate greater than any other cancer. To systematically search for new genes involved in melanomagenesis, we collated exome sequencing data from independent melanoma cohort datasets, including those in the public domain. We identified recurrent mutations that may drive melanoma growth, survival or metastasis, and which may hold promise for the design of novel therapies to treat melanoma. These included a frequent recurrent (i.e. hotspot) mutation in the 5' untranslated region of RPS27 in ~10% of samples. We show that the mutation expands the 5'TOP element, a motif known to regulate the expression of most of the ribosomal protein family, to which RPS27 belongs, and thus might sensitize the mutated transcript to growth-mediated regulation. This finding highlights not only the important role of non-protein coding genetic aberrations in cancer development but also their potential as novel therapeutic targets.
Martin D., Abba M., Molinolo A., Vitale-Cross L., Wang Z., Zaida M., Delic N., Samuels Y., Lyons J. & Gutkind J.
(2014)
Oncotarget.
5,
19,
p. 8906-8923
The recent elucidation of the genomic landscape of head and neck squamous cell carcinoma (HNSCC) has provided a unique opportunity to develop selective cancer treatment options. These efforts will require the establishment of relevant HNSCC models for preclinical testing. Here, we performed full exome and transcriptome sequencing of a large panel of HNSCC-derived cells from different anatomical locations and human papillomavirus (HPV) infection status. These cells exhibit typical mutations in TP53, FAT1, CDK2NA, CASP8, and NOTCH1, and copy number variations (CNVs) and mutations in PIK3CA, HRAS, and PTEN that reflect the widespread activation of the PI3K-mTOR pathway. SMAD4 alterations were observed that may explain the decreased tumor suppressive effect of TGF-β in HNSCC. Surprisingly, we identified HPV+ HNSCC cells harboring TP53 mutations, and documented aberrant TP53 expression in a subset of HPV+ HNSCC cases. This analysis also revealed that most HNSCC cells harbor multiple mutations and CNVs in epigenetic modifiers (e.g., EP300, CREBP, MLL1, MLL2, MLL3, KDM6A, and KDM6B) that may contribute to HNSCC initiation and progression. These genetically-defined experimental HNSCC cellular systems, together with the identification of novel actionable molecular targets, may now facilitate the pre-clinical evaluation of emerging therapeutic agents in tumors exhibiting each precise genomic alteration.
Lau C., Killian K. J., Samuels Y. & Rudloff U.
(2013)
Molecular Diagnostics For Melanoma
: Methods And Protocols
.
Marincola F. M. & Thurin M.(eds.).
p. 461-480
Recent sequencing efforts in melanoma have elucidated many previously unknown molecular pathways and biological mechanisms involved in melanoma development and progression and have yielded a number of promising targets for molecular therapy. As sequencing technologies have become more sophisticated and have revealed an ever-increasing complexity of the genetic landscape of melanoma, it has become clear that sequencing methods applied to clinical specimens have to reliably capture not only recurrent "hotspot" mutations like BRAFV600 and NRASQ61 or "mini-hotspot" mutations like exon 11 and 13 c-KIT but also heterogeneous somatic mutations dispersed across multiple functionally conserved regions of genes or entire genes. One such example in melanoma is the ERBB4 receptor, or HER4, a member of the Erb receptor family, which has recently been shown to be a major oncogenic "driver" in melanoma. Mutated ERBB4 signaling activates both aberrant ERBB4 and PI3K-AKT signal transduction, mediates sensitivity to small-molecule inhibition with the dual-tyrosine kinase inhibitor lapatinib, and has recently also been implied in oncogenic glutamatergic signaling in melanoma. Mutations involving the ERBB4 gene act as "gain-of-function" mutations and predominantly involve the extracellular domains of the receptor. Additional sequencing efforts have recently identified recurrent mutations ("mini-hotspots") or mutation clusters which affect the regulation of, e. g., ligand binding, arrangement of extracellular domain alignment, or intramolecular tether formation. In this chapter, we describe the methods used to determine the mutation status of all exons of the ERBB4 gene in clinical specimens obtained from patients afflicted by metastatic melanoma. Upon slight modifications, this protocol can also be used for mutational analysis of other oncogenes affected by "non-hotspot" mutations dispersed across multiple exons. This sequencing technique has successfully been applied within a clinical trial selecting patients with ERBB4-mutant melanoma for lapatinib treatment. With the increasing emergence of low-frequency oncogenes affected by heterogeneous activating mutations located in different exons and regions this method will provide a mean to translate the promise of recently obtained genetic knowledge into clinical genotype-directed targeted therapy trials.
Gartner J. J., Parker S. C., Prickett T. D., Dutton-Regester K., Stitzel M. L., Lin J. C., Davis S., Simhadri V. L., Jha S., Katagiri N., Gotea V., Teer J. K., Wei X., Morken M. A., Bhanot U. K., Chen G., Elnitski L. L., Davies M. A., Gershenwald J. E., Carter H., Karchin R., Robinson W., Robinson S., Rosenberg S. A., Collins F. S., Parmigiani G., Komar A. A., Kimchi-Sarfaty C., Hayward N. K., Margulies E. H. & Samuels Y.
(2013)
Proceedings of the National Academy of Sciences of the United States of America.
110,
33,
p. 13481-13486
Synonymous mutations, which do not alter the protein sequence, have been shown to affect protein function [Sauna ZE, Kimchi- Sarfaty C (2011) Nat Rev Genet 12(10):683-691]. However, synonymous mutations are rarely investigated in the cancer genomics field. We used whole-genome and -exome sequencing to identify somatic mutations in 29 melanoma samples. Validation of one synonymous somatic mutation in BCL2L12 in 285 samples identified 12 cases that harbored the recurrent F17F mutation. This mutation led to increased BCL2L12 mRNA and protein levels because of differential targeting of WT and mutant BCL2L12 by hsa-miR- 671-5p. Protein made from mutant BCL2L12 transcript bound p53, inhibited UV-induced apoptosis more efficiently than WT BCL2L12, and reduced endogenous p53 target gene transcription. This report shows selection of a recurrent somatic synonymous mutation in cancer. Our data indicate that silent alterations have a role to play in human cancer, emphasizing the importance of their investigation in future cancer genome studies.
Lu Y. C., Yao X., Li Y. F., El-Gamil M., Dudley M. E., Yang J. C., Almeida J. R., Douek D. C., Samuels Y., Rosenberg S. A. & Robbins P. F.
(2013)
Journal of Immunology.
190,
12,
p. 6034-6042
Adoptive cell therapy with tumor-infiltrating lymphocytes (TILs) represents an effective treatment for patients with metastatic melanoma. However, most of the Ag targets recognized by effective melanoma-reactive TILs remain elusive. In this study, patient 2369 experienced a complete response, including regressions of bulky liver tumor masses, ongoing beyond 7 y following adoptive TIL transfer. The screening of a cDNA library generated from the autologous melanoma cell line resulted in the isolation of a mutated protein phosphatase 1, regulatory (inhibitor) subunit 3B (PPP1R3B) gene product. The mutated PPP1R3B peptide represents the immunodominant epitope recognized by tumor-reactive T cells in TIL 2369. Five years following adoptive transfer, peripheral blood T lymphocytes obtained from patient 2369 recognized the mutated PPP1R3B epitope. These results demonstrate that adoptive T cell herapy targeting a tumor-specific Ag can mediate long-term survival for a patient with metastatic melanoma. This study also provides an impetus to develop personalized immunotherapy targeting tumor-specific, mutated Ags.
Hansen N. F., Gartner J. J., Mei L., Samuels Y. & Mullikin J. C.
(2013)
Bioinformatics.
29,
12,
p. 1498-1503
Motivation: Extensive DNA sequencing of tumor and matched normal samples using exome and whole-genome sequencing technologies has enabled the discovery of recurrent genetic alterations in cancer cells, but variability in stromal contamination and subclonal heterogeneity still present a severe challenge to available detection algorithms.Results: Here, we describe publicly available software, Shimmer, which accurately detects somatic single-nucleotide variants using statistical hypothesis testing with multiple testing correction. This program produces somatic single-nucleotide variant predictions with significantly higher sensitivity and accuracy than other available software when run on highly contaminated or heterogeneous samples, and it gives comparable sensitivity and accuracy when run on samples of high purity.
Robbins P. F., Lu Y., El-Gamil M., Li Y. F., Gross C., Gartner J., Lin J. C., Teer J. K., Cliften P., Tycksen E., Samuels Y. & Rosenberg S. A.
(2013)
Nature Medicine.
19,
6,
p. 747-752
Substantial regressions of metastatic lesions have been observed in up to 70% of patients with melanoma who received adoptively transferred autologous tumor-infiltrating lymphocytes (TILs) in phase 2 clinical trials. In addition, 40% of patients treated in a recent trial experienced complete regressions of all measurable lesions for at least 5 years following TIL treatment. To evaluate the potential association between the ability of TILs to mediate durable regressions and their ability to recognize potent antigens that presumably include mutated gene products, we developed a new screening approach involving mining whole-exome sequence data to identify mutated proteins expressed in patient tumors. We then synthesized and evaluated candidate mutated T cell epitopes that were identified using a major histocompatibility complex-binding algorithm for recognition by TILs. Using this approach, we identified mutated antigens expressed on autologous tumor cells that were recognized by three bulk TIL lines from three individuals with melanoma that were associated with objective tumor regressions following adoptive transfer. This simplified approach for identifying mutated antigens recognized by T cells avoids the need to generate and laboriously screen cDNA libraries from tumors and may represent a generally applicable method for identifying mutated antigens expressed in a variety of tumor types.
Prickett T., Hill V., Gartner J., Zerlanko B., Jiang J., Samaan M., Wunderlich J., Gutkind S., Rosenberg S. & Samuels Y.
(2013)
Cancer Research.
73,
8,
Hill V. K., Gartner J. J., Samuels Y. & Goldstein A. M.
(2013)
Annual Review of Genomics and Human Genetics.
14,
p. 257-279
Cutaneous malignant melanoma results from the interplay of genetic, host, and environmental factors. Genetic factors implicated in melanoma etiology include inherited high-, intermediate-, and low-risk susceptibility genes as well as numerous somatic mutations in melanoma tumors. CDKN2A is the major high-risk melanoma susceptibility gene identified to date. Recent identification of low-risk loci has been accomplished predominantly through genome-wide association studies. Whole-exome and whole-genome studies have identified numerous genes somatically altered in melanoma tumors and highlighted a higher mutation load in melanoma tumors compared with those in other cancers. This higher load is believed to be attributable to the preponderance of cytosine-to-thymine nucleotide substitutions as a result of UV radiation exposure. Technological advances, particularly next-generation sequencing, have increased the opportunities for germline and somatic gene discovery in melanoma and are opening up new avenues for understanding melanoma pathogenesis as well as leading to new opportunities for treatment.
Gartner J. J., Davis S., Wei X., Lin J. C., Trivedi N. S., Teer J. K., Meltzer P. S., Rosenberg S. A. & Samuels Y.
(2012)
BMC Genomics.
13,
1,
505.
Background: Metastasis is characterized by spreading of neoplastic cells to an organ other than where they originated and is the predominant cause of death among cancer patients. This holds true for melanoma, whose incidence is increasing more rapidly than any other cancer and once disseminated has few therapeutic options. Here we performed whole exome sequencing of two sets of matched normal and metastatic tumor DNAs.Results: Using stringent criteria, we evaluated the similarities and differences between the lesions. We find that in both cases, 96% of the single nucleotide variants are shared between the two metastases indicating that clonal populations gave rise to the distant metastases. Analysis of copy number variation patterns of both metastatic sets revealed a trend similar to that seen with our single nucleotide variants. Analysis of pathway enrichment on tumor sets shows commonly mutated pathways enriched between individual sets of metastases and all metastases combined.Conclusions: These data provide a proof-of-concept suggesting that individual metastases may have sufficient similarity for successful targeting of driver mutations.
Prickett T. & Samuels Y.
(2012)
Clinical Cancer Research.
18,
16,
p. 4240-4246
The neurotransmitter glutamate interacts with glutamate receptor proteins, leading to the activation of multiple signaling pathways. Dysfunction in the glutamatergic signaling pathway is well established as a frequent player in diseases such as schizophrenia, Alzheimer disease, and brain tumors (gliomas). Recently, aberrant functioning of this pathway has also been shown in melanoma. In both glioma and melanoma, glutamate secretion stimulates tumor growth, proliferation, and survival through activation of the mitogen-activated protein kinase and phosphoinositide 3-kinase/Akt pathways. In the future, extracellular glutamate levels and glutamatergic signaling may serve as biological markers for tumorigenicity and facilitate targeted therapy for melanoma.
Walia V., Mu E. W., Lin J. C. & Samuels Y.
(2012)
Pigment Cell and Melanoma Research.
25,
2,
p. 155-170
Melanoma, the most aggressive form of skin cancer, has increased in incidence more rapidly than any other cancer. The completion of the human genome project and advancements in genomics technologies has allowed us to investigate genetic alterations of melanoma at a scale and depth that is unprecedented. Here, we survey the history of the different approaches taken to understand the genomics of melanoma - from early candidate genes, to gene families, to genome-wide studies. The new era of whole-exome and whole-genome sequencing has paved the way for an in-depth understanding of melanoma biology, identification of new therapeutic targets, and development of novel personalized therapies for melanoma. Published 2012. This article is a U.S. Government work and is in the public domain in the USA.
Prickett T., Wei X., Cardenas-Navia I., Teer J., Lin J., Walia V., Gartner J., Jiang J., Cherukuri P., Molinolo A., Davies M., Gershenwald J., Stemke-Hale K., Rosenberg S., Margulies E. & Samuels Y.
(2011)
Nature Genetics.
43,
11,
p. 1119-1126
G protein-coupled receptors (GPCRs), the largest human gene family, are important regulators of signaling pathways. However, knowledge of their genetic alterations is limited. In this study, we used exon capture and massively parallel sequencing methods to analyze the mutational status of 734 GPCRs in melanoma. This investigation revealed that one family member, GRM3, was frequently mutated and that one of its mutations clustered within one position. Biochemical analysis of GRM3 alterations revealed that mutant GRM3 selectively regulated the phosphorylation of MEK, leading to increased anchorage-independent growth and migration. Melanoma cells expressing mutant GRM3 had reduced cell growth and cellular migration after short hairpin RNAĝ\u20ac"mediated knockdown of GRM3 or treatment with a selective MEK inhibitor, AZD-6244, which is currently being used in phase 2 clinical trials. Our study yields the most comprehensive map of genetic alterations in the GPCR gene family.
Mutations of the PIK3CA gene in human cancers
Samuels Y., Velculescu V., Kinzler K. W. & Vogelstein B.
(2011)
02 Mar 2004,
Phosphatidylinositol 3-kinases (PI3Ks) are known to be important regulators of signaling pathways. To determine whether PI3Ks are genetically altered in cancers, we analyzed the sequences of the P13K gene family and discovered that one family member, PIK3CA, is frequently mutated in cancers of the colon and other organs. The majority of mutations clustered near two positions within the P13K helical or kinase domains. PIK3CA represents one of the most highly mutated oncogenes yet identified in human cancers and is useful as a diagnostic and therapeutic target.
Solomon D., Kim T., Diaz-Martinez L., Fair J., Elkahloun A., Harris B., Toretsky J., Rosenberg S., Shukla N., Ladanyi M., Samuels Y., James C., Yu H., Kim J. & Waldman T.
(2011)
Science.
333,
6045,
p. 1039-1043
Most cancer cells are characterized by aneuploidy, an abnormal number of chromosomes. We have identified a clue to the mechanistic origins of aneuploidy through integrative genomic analyses of human tumors. A diverse range of tumor types were found to harbor deletions or inactivating mutations of STAG2, a gene encoding a subunit of the cohesin complex, which regulates the separation of sister chromatids during cell division. Because STAG2 is on the X chromosome, its inactivation requires only a single mutational event. Studying a near-diploid human cell line with a stable karyotype, we found that targeted inactivation of STAG2 led to chromatid cohesion defects and aneuploidy, whereas in two aneuploid human glioblastoma cell lines, targeted correction of the endogenous mutant alleles of STAG2 led to enhanced chromosomal stability. Thus, genetic disruption of cohesin is a cause of aneuploidy in human cancer.
Wei X., Moncada-Pazos A., Cal S., Soria-Valles C., Gartner J., Rudloff U., Lin J., Rosenberg S., Lopez-Otin C. & Samuels Y.
(2011)
Human Mutation.
32,
6,
p. E2148-E2175
We performed a mutational analysis of the 19 disintegrin-metalloproteinases (ADAMs) genes in human cutaneous metastatic melanoma and identified eight to be somatically mutated in 79 samples, affecting 34% of the melanoma tumors analyzed. Functional analysis of the two frequently mutated ADAM genes, ADAM29 and ADAM7 demonstrated that the mutations affect adhesion of melanoma cells to specific extracellular matrix proteins and in some cases increase their migration ability. This suggests that mutated ADAM genes could play a role in melanoma progression.
Wei X., Walia V., Lin J., Teer J., Prickett T., Gartner J., Davis S., Stemke-Hale K., Davies M., Gershenwald J., Robinson W., Robinson S., Rosenberg S. & Samuels Y.
(2011)
Nature Genetics.
43,
5,
p. 442-446
The incidence of melanoma is increasing more than any other cancer, and knowledge of its genetic alterations is limited. To systematically analyze such alterations, we performed whole-exome sequencing of 14 matched normal and metastatic tumor DNAs. Using stringent criteria, we identified 68 genes that appeared to be somatically mutated at elevated frequency, many of which are not known to be genetically altered in tumors. Most importantly, we discovered that TRRAP harbored a recurrent mutation that clustered in one position (p. Ser722Phe) in 6 out of 167 affected individuals (∼1/44%), as well as a previously unidentified gene, GRIN2A, which was mutated in 33% of melanoma samples. The nature, pattern and functional evaluation of the TRRAP recurrent mutation suggest that TRRAP functions as an oncogene. Our study provides, to our knowledge, the most comprehensive map of genetic alterations in melanoma to date and suggests that the glutamate signaling pathway is involved in this disease.
Samuels Y. & Waldman T.
(2011)
PHOSPHOINOSITIDE 3-KINASE IN HEALTH AND DISEASE, VOL 2
.
p. 21-41
(trueCurrent Topics in Microbiology and Immunology).
The involvement of the PIK3CA gene product p110 alpha, the catalytic subunit of phosphatidylinositol 3-kinase (PI3K), in human cancer has been suggested for over 15 years, and support for this proposal had been provided by both genetic and functional studies, including most recently the discovery of common activating missense mutations of PIK3CA in a wide variety of common human tumor types. This chapter will focus on the discovery of these mutations and describes their relevance to a wide range of common human tumor types. Of note, the identification and functional analysis of the PIK3CA gene are reviewed in other chapters in this book. However, a brief mention will be made here of its general properties as background to our focus on the discovery of its cancer-specific mutations.
Wei X., Prickett T., Viloria C., Molinolo A., Lin J., Cardenas-Navia I., Cruz P., Rosenberg S., Davies M., Gershenwald J., Lopez-Otin C. & Samuels Y.
(2010)
Molecular Cancer Research.
8,
11,
p. 1513-1525
The disintegrin-metalloproteinases with thrombospondin domains (ADAMTS) genes have been suggested to function as tumor suppressors as several have been found to be epigenetically silenced in various cancers. We performed a mutational analysis of the ADAMTS gene family in human melanoma and identified a large fraction of melanomas to harbor somatic mutations. To evaluate the functional consequences of the most commonly mutated gene, ADAMTS18, six of its mutations were biologically examined. ADAMTS18 mutations had little effect on melanoma cell growth under standard conditions, but reduced cell dependence on growth factors. ADAMTS18 mutations also reduced adhesion to laminin and increased migration in vitro and metastasis in vivo. Melanoma cells expressing mutant ADAMTS18 had reduced cell migration after short hairpin RNA-mediated knockdown of ADAMTS18, suggesting that ADAMTS18 mutations promote growth, migration, and metastasis in melanoma.
Cardenas-Navia L., Cruz P., Lin J., Rosenberg S. & Samuels Y.
(2010)
Cancer Biology & Therapy.
10,
1,
p. 33-37
Heterotrimeric guanine nucleotide-binding proteins (G proteins) mediate signals between G-protein coupled receptors and their downstream pathways, and have been shown to be mutated in cancer. In particular, GNAQ was found to be frequently mutated in blue nevi of the skin and uveal melanoma, acting as an oncogene in its mutated form. To further examine the role of heterotrimeric G proteins in melanoma, we performed a comprehensive mutational analysis of the 35 genes in the heterotrimeric G protein gene family in a panel of 80 melanoma samples. somatic alterations in a G protein subunit were detected in 17% of samples spanning 7 genes. The highest rates of somatic, non-synonymous mutations were found in GNG10 and GNAZ, neither of which has been previously reported to be mutated in melanoma. Our study is the first systematic analysis of the heterotrimeric G proteins in melanoma and indicates that multiple mutated heterotrimeric G proteins may be involved in melanoma progression.
Lopez G., Reitman Z., Solomon D., Waldman T., Bigner D., McLendon R., Rosenberg S., Samuels Y. & Yan H.
(2010)
Biochemical and Biophysical Research Communications.
398,
3,
p. 585-587
Isocitrate dehydrogenase 1 (IDH1) and isocitrate dehydrogenase 2 (IDH2) are enzymes which convert isocitrate to α-ketoglutarate while reducing nicotinamide adenine dinucleotide phosphate (NADP. +. to NADPH). IDH1/2 were recently identified as mutated in a large percentage of progressive gliomas. These mutations occur at IDH1R132 or the homologous IDH2R172. Melanomas share some genetic features with IDH1/2-mutated gliomas, such as frequent TP53 mutation. We sought to test whether melanoma is associated with IDH1/2 mutations. Seventy-eight human melanoma samples were analyzed for IDH1R132 and IDH2R172 mutation status. A somatic, heterozygous IDH1 c.C394T (p.R132C) mutation was identified in one human melanoma metastasis to the lung. Having identified this mutation in one metastasis, we sought to test the hypothesis that certain selective pressures in the brain environment may specifically favor the cell growth or survival of tumor cells with mutations in IDH1/2, regardless of primary tumor site. To address this, we analyzed IDH1R132 and IDH2R172 mutation status 53 metastatic brain tumors, including nine melanoma metastases. Results revealed no mutations in any samples. This lack of mutations would suggest that mutations in IDH1R132 or IDH2R172 may be necessary for the formation of tumors in a cell-lineage dependent manner, with a particularly strong selective pressure for mutations in progressive gliomas; this also suggests the lack of a particular selective pressure for growth in brain tissue in general. Studies on the cell-lineages of tumors with IDH1/2 mutations may help clarify the role of these mutations in the development of brain tumors.
Rudloff U. & Samuels Y.
(2010)
Cell Cycle.
9,
8,
p. 1487-1503
As the upward spiral of novel cancer gene discoveries and novel molecular compounds continues to accelerate, a repetitive theme in molecular drug development remains the lack of activity of initially promising agents when given to patients in clinical trials. It is however invigorating that a few targeted agents directed against a select group of a few 'cancer gene superfamilies' have escaped this all to common fate, and have evolved into novel, clinically meaningful molecular therapy strategies. Targeting dysregulated signaling of the epidermal growth factor family of transmembrane receptors (Erbb family) has encompassed over the last decade an ever increasing role in personalized treatment approaches in an increasing number of human malignancies. Erbbs are receptor tyrosine kinases that are important regulators of several signaling pathways. Two of its family members (Erbb1/EGFR and Erbb2/HER2) have previously been shown to be somatically mutated of human cancers. To determine if this family is somatically mutated in melanoma, its sequences were recently analyzed and one of its members, Erbb4, was found to be somatically mutated in 19% of melanoma cases. Functional analysis of seven of its mutations was shown to increase its catalytic and transformation abilities as well as providing essential survival signals. Similar to other Erbb family members, mutant Erbb4 seems to confer "oncogene addiction" on melanoma cells, making it an attractive therapeutic target. Gaining further understanding into the oncogenic mechanism of Erbb4 may not only help in the development of targeted therapy in melanoma patients but might accelerate the acceptance of a novel taxonomy of cancer which is based on the genomic perturbations in cancer genes and cancer gene families and their response to targeted agents.
Ericson K., Gan C., Cheong I., Rago C., Samuels Y., Velculescu V. E., Kinzler K. W., Huso D. L., Vogelstein B. & Papadopoulos N.
(2010)
Proceedings of the National Academy of Sciences of the United States of America.
107,
14,
p. 6551
Lopez-Otin C., Palavalli L. H. & Samuels Y.
(2009)
Cell Cycle.
8,
22,
p. 3657-3662
Matrix metalloproteinases (MMPs) have long been linked to cancer progression owing to their ability to breakdown tissue barriers for metastatic spread. Accordingly, multiple studies have examined the potential value of these enzymes as targets for cancer therapy. Unfortunately, most clinical trials with MMP inhibitors have yielded negative results which has made necessary to re-evaluate the role of these proteases in cancer. Recent works mainly based on the use of mouse models deficient in specific MMPs have revealed that these enzymes play many roles in cancer distinct from matrix destruction, influencing early steps of tumor evolution, and expanding their pro-tumorigenic properties. However, these in vivo studies have also shown that, unexpectedly, some MMP family members like MMP8 may have paradoxical anti-tumor functions. Nevertheless, the final validation of these MMPs as bona fide tumor suppressors requested the identification of the putative genetic or epigenetic changes underlying their inactivation during cancer development. To this purpose, very recent large-scale genomic studies have explored the possibility that MMPs could be genetically altered in a panel of human malignant tumors from different sources. These studies have demonstrated that MMP8 is a frequently mutated gene in human melanoma. Functional analysis of the identified mutations has confirmed that all of them lead to the loss-of-function of MMP8 and enhance the progression of melanoma, thus providing definitive evidence that MMP8 is a tumor-suppressor gene. Parallel studies have extended these findings to other MMP-related metalloproteinases such as ADAMTS15, which has been found to be genetically inactivated in human colorectal cancer. This review describes the identification and validation of some MMPs and related enzymes as anti-tumor proteases and speculates about the molecular mechanisms underlying their protective roles in tumor development. Finally, the review explores the clinical applications derived from the identification of MMPs that favour the host instead of the tumor.
Cronin J., Wunderlich J., Loftus S., Prickett T., Wei X., Ridd K., Vemula S., Burrell A., Agrawal N., Lin J., Banister C., Buckhaults P., Rosenberg S., Bastian B., Pavan W. & Samuels Y.
(2009)
Pigment Cell and Melanoma Research.
22,
4,
p. 435-444
Microphthalmia-associated transcription factor (MITF) is involved in melanocyte cell development, pigmentation and neoplasia. To determine whether MITF is somatically mutated in melanoma, we compared the sequence of MITF from primary and metastatic lesions to patient-matched normal DNA. In the 50 metastatic melanoma tumor lines analysed, we discovered four samples that had genomic amplifications of MITF and four that had MITF mutations in the regions encoding the transactivation, DNA binding or basic, helix-loop-helix domains. Sequence analysis for SOX10, a transcription factor, which both acts upstream of MITF and synergizes with MITF, identified an additional three samples with frameshift or nonsense mutations. Microphthalmia-associated transcription factor and SOX10 were found to be mutated in a mutually exclusive fashion, possibly suggesting disruption in a common genetic pathway. Taken together we found that over 20% of the metastatic melanoma cases had alterations in the MITF pathway. We show that the MITF pathway is also altered in primary melanomas: 2/26 demonstrated mutations in MITF and 6/55 demonstrated mutations in SOX10. Our findings suggest that altered MITF function during melanomagenesis can be achieved by MITF amplification, MITF single base substitutions or by mutation of its regulator SOX10.
Palavalli L., Prickett T., Wunderlich J., Wei X., Burrell A., Porter-Gill P., Davis S., Wang C., Cronin J., Agrawal N., Lin J., Westbroek W., Hoogstraten-Miller S., Molinolo A., Fetsch P., Filie A., O'Connell M., Banister C., Howard J., Buckhaults P., Weeraratna A., Brody L., Rosenberg S. & Samuels Y.
(2009)
Nature Genetics.
41,
5,
p. 518-520
A mutational analysis of the matrix metalloproteinase (MMP) gene family in human melanoma identified somatic mutations in 23% of melanomas. Five mutations in one of the most commonly mutated genes, MMP8, reduced MMP enzyme activity. Expression of wild-type but not mutant MMP8 in human melanoma cells inhibited growth on soft agar in vitro and tumor formation in vivo, suggesting that wild-type MMP-8 has the ability to inhibit melanoma progression.
Solomon D. A., Kim J. S., Cronin J. C., Sibenaller Z., Ryken T., Rosenberg S. A., Ressom H., Jean W., Bigner D., Yan H., Samuels Y. & Waldman T.
(2008)
Cancer Research.
68,
24,
p. 10300-10306
An additional tumor suppressor gene on chromosome 9p telomeric to the CDKN2A/B locus has long been postulated to exist. Using Affymetrix 250K single nucleotide polymorphism arrays to screen for copy number changes in glioblastoma multiforme (GBM), we detected a high frequency of deletions of the PTPRD gene, which encodes a receptor protein tyrosine phosphatase at chromosome 9p23-24.1. Missense and nonsense mutations of PTPRD were identified in a subset of the samples lacking deletions, including an inherited mutation with somatic loss of the wild-type allele. We then sequenced the gene in melanoma and identified 10 somatic mutations in 7 of 57 tumors (12%). Reconstitution of PTPRD expression in GBM and melanoma cells harboring deletions or mutations led to growth suppression and apoptosis that was alleviated by both the somatic and constitutional mutations. These data implicate PTPRD in the pathogenesis of tumors of neuroectodermal origin and, when taken together with other recent reports of PTPRD mutations in adenocarcinoma of the colon and lung, suggest that PTPRD may be one of a select group of tumor suppressor genes that are inactivated in a wide range of common human tumor types.
Huang C., Mandelker D., Schmidt-Kittler O., Samuels Y., Velculescu V., Kinzler K., Vogelstein B., Gabelli S. & Amzel L.
(2007)
Science.
318,
5857,
p. 1744-1748
PIK3CA, one of the two most frequently mutated oncogenes in human tumors, codes for p110α, the catalytic subunit of a phosphatidylinositol 3-kinase, isoform α (PI3Kα, p110α/p85). Here, we report a 3.0 angstrom resolution structure of a complex between p110α and a polypeptide containing the p110α-binding domains of p85α, a protein required for its enzymatic activity. The structure shows that many of the mutations occur at residues lying at the interfaces between p110α and p85α or between the kinase domain of p110α and other domains within the catalytic subunit. Disruptions of these interactions are likely to affect the regulation of kinase activity by p85 or the catalytic activity of the enzyme, respectively. In addition to providing new insights about the structure of PI3Kα, these results suggest specific mechanisms for the effect of oncogenic mutations in p110α and p85α.
Rago C., Huso D., Diehl F., Karim B., Liu G., Papadopoulos N., Samuels Y., Velculescu V., Vogelstein B., Kinzler K. & Diaz L.
(2007)
Cancer Research.
67,
19,
p. 9364-9370
Internal human xenografts provide valuable animal models to study the microenvironments and metastatic processes occurring in human cancers. However, the use of such models is hampered by the logistical difficulties of reproducibly and simply assessing tumor burden. We developed a high-sensitivity assay for quantifying human DNA in small volumes of mouse plasma, enabling in-life monitoring of systemic tumor burden. Growth kinetics analyses of various xenograft models showed the utility of circulating human DNA as a biomarker. We found that human DNA concentration reproducibly increased with disease progression and decreased after successful therapeutic intervention. A marked, transient spike in circulating human tumor DNA occurred immediately after cytotoxic therapy or surgery. This simple assay may find broad utility in target validation studies and preclinical drug development programs.
Bergamaschi D., Samuels Y., Sullivan A., Zvelebil M., Breyssens H., Bisso A., Del Sal S. G., Syed N., Smith P., Gasco M., Crook T. & Lu X.
(2006)
Nature Genetics.
38,
10,
p. 1133-1141
iASPP is one of the most evolutionarily conserved inhibitors of p53, whereas ASPP1 and ASPP2 are activators of p53. We show here that, in addition to the DNA-binding domain, the ASPP family members also bind to the proline-rich region of p53, which contains the most common p53 polymorphism at codon 72. Furthermore, the ASPP family members, particularly iASPP, bind to and regulate the activity of p53Pro72 more efficiently than that of p53Arg72. Hence, escape from negative regulation by iASPP is a newly identified mechanism by which p53Arg72 activates apoptosis more efficiently than p53Pro72.
Nakayama K., Nakayama N., Kurman R., Cope L., Pohl G., Samuels Y., Velculescu V., Wang T. & Shih L.
(2006)
Cancer Biology & Therapy.
5,
7,
p. 779-785
Sequence mutations and gene amplifications lead to activation of the PIK3CA-AKT2 signaling pathway and have been reported in several types of neoplasms including ovarian cancer. Analysis of such genetic alterations, however, is usually complicated by contamination of normal cell DNA, artifacts associated with formalin-fixed tissues and the sensitivity of the techniques employed. In this study, we analyzed the sequence mutations in PIK3CA and AKT2 genes using purified tumor cells that were isolated from high-grade ovarian serous carcinomas and serous borderline tumors (SBTs) and assessed gene amplification using a dual-color FISH on tissue microarrays. Somatic sequence mutations in the kinase domain of AKT2 were not detected in any of the 65 ovarian tumors analyzed. Mutations of PIK3CA were rare, occurring only in one (2.3%) of 44 high-grade serous carcinomas and in only one (4.8%) of 21 SBTs. Dual-color FISH demonstrated that PIK3CA and AKT2 were not amplified in SBTs but amplified in 13.3% and 18.2% high-grade carcinomas, respectively. High-level amplification (>3 fold) was more frequently observed in AKT2 than in PIK3CA. Unlike mutations in ERBB2, KRAS and BRAF which are mutually exclusive in SBTs, coamplification of PIK3CA and AKT2 was present in five high-grade carcinomas including the OVCAR3 cells. Amplification in either of the genes occurred in 27% high-grade serous carcinomas. In conclusion, the methods we employed provide unambiguous evidence that somatic sequence mutations of PIK3CA and ATK2 are rare in ovarian serous tumors but amplification of both genes may play an important role in the development of high-grade ovarian serous carcinoma.
Samuels Y. & Ericson K.
(2006)
Current Opinion in Oncology.
18,
1,
p. 77-82
Purpose of review: The purpose of this review is to examine the contribution of the PI3K signaling pathway to the development of human tumors and to propose further studies to elucidate how to develop therapeutics for patients with mutations in this pathway. Recent findings: More than 30% of various solid tumor types were recently found to contain mutations in PIK3CA, the catalytic subunit of PI3K. Further analysis of key genes in this pathway identified an additional eight genes altered in tumors. These were generally found to be mutated in a mutually exclusive manner, thus increasing the mutation frequency of the pathway to 40% in colorectal cancers and emphasizing the importance of the PI3K pathway in tumorigenesis. Functional analyses of PIK3CA mutations revealed that they increase its enzymatic activity, stimulate AKT signaling, allow growth factor-independent growth as well as increasing cell invasion and metastasis. Summary: The PI3K signaling pathway is dysregulated by a variety of mechanisms in a large fraction of human tumors. Both mutational and functional analyses have shown that PIK3CA is an oncogene that plays an important role in tumor progression. Mutant members of the PI3K pathway, including PIK3CA, are good targets for therapeutic intervention because most of them are kinases, making them attractive for drug development. Gaining further insights into PIK3CA oncogenic mechanisms may produce new biomarkers and help the development of targeted therapeutics.
Samuels Y., Diaz L., Schmidt-Kittler O., Cummins J., DeLong L., Cheong I., Rago C., Huso D., Lengauer C., Kinzler K., Vogelstein B. & Velculescu V.
(2005)
Cancer Cell.
7,
6,
p. 561-573
PIK3CA is mutated in diverse human cancers, but the functional effects of these mutations have not been defined. To evaluate the consequences of PIK3CA alterations, the two most common mutations were inactivated by gene targeting in colorectal cancer (CRC) cells. Biochemical analyses of these cells showed that mutant PIK3CA selectively regulated the phosphorylation of AKT and the forkhead transcription factors FKHR and FKHRL1. PIK3CA mutations had little effect on growth under standard conditions, but reduced cellular dependence on growth factors. PIK3CA mutations resulted in attenuation of apoptosis and facilitated tumor invasion. Treatment with the PI3K inhibitor LY294002 abrogated PIK3CA signaling and preferentially inhibited growth of PIK3CA mutant cells. These data have important implications for therapy of cancers harboring PIK3CA alterations.
Bergamaschi D., Samuels Y., Zhong S. & Lu X.
(2005)
Oncogene.
24,
23,
p. 3836-3841
Using various mutants of p53 and mdm2, we demonstrate here that both the DNA binding and transactivation function of p53 are required for ASPP1 and ASPP2 to stimulate the apoptotic functions of p53. Mdm2 and mdmx prevent ASPP1 and ASPP2 from stimulating the apoptotic function of p53 by binding and inhibiting the transcriptional activity of p53. Importantly, mdm2 and mdmx can prevent the stimulatory effects of ASPP1 and ASPP2 without targeting p53 for degradation. These data provide a novel mechanism by which mdm2 and mdmx act as potent inhibitors of p53.
Samuels Y., Zhu J. & Lengauer C.
(2005)
Cancer Biology and Therapy.
4,
5,
p. 546-547
Commentary to "PWT-458, A Novel Pegylated-17-Hydroxywortmannin, Inhibits Phosphatidylinositol 3-Kinase Signaling and Suppresses Growth of Solid Tumors"
Erratum: The PIK3CA gene is mutated with high frequency in human breast cancers (Cancer Biology and Therapy (2004) 3 (772-775))
Lin L., Bachman K. E., Ellis M., Ben H. P., Bachman K. E., Argani P., Samuels Y., Silliman N., Ptak J., Szabo S., Konishi H., Karakas B., Blair B. G., Lin C., Peters B. A., Velculescu V. E. & Park B. H.
(2005)
Cancer Biology and Therapy.
4,
2,
p. 133
Samuels Y. & Velculescu V.
(2004)
Cell Cycle.
3,
10,
p. 1221-1224
Phosphatidylinositol 3-kinases (PI3Ks) are important regulators of signaling pathways. To determine whether PI3Ks are genetically altered in human cancers, we recently analyzed the sequences of the PI3K gene family and discovered that one member, the PIK3CA gene encoding the p110α catalytic subunit, was frequently mutated in cancers of the colon, breast, brain and lung. The majority of mutations clustered near two positions within the PI3K helical or catalytic domains and at least one hotspot mutation appeared to increase kinase activity. PIK3CA represents one of the most highly mutated oncogenes identified in human cancers and may be a useful diagnostic and therapeutic target.
Bachman K., Argani P., Samuels Y., Silliman N., Ptak J., Szabo S., Konishi H., Karakas B., Blair B., Lin C., Peters B., Velculescu V. & Park B.
(2004)
Cancer Biology and Therapy.
3,
8,
p. 772-775
The phosphatidylinositol 3-kinases (PI3Ks) are known regulators of cellular growth and proliferation. It has recently been reported that somatic mutations within the PI3K subunit p110α (PIK3CA) are present in human colorectal and other cancers. Here we show that thirteen of fifty-three breast cancers (25%) contain somatic mutations in PIK3CA, with the majority of mutations located in the kinase domain. These results demonstrate that PIK3CA is the most mutated oncogene in breast cancer and support a role for PIK3CA in epithelial carcinogenesis.
Samuels Y., Wang Z., Bardelli A., Silliman N., Ptak J., Szabo S., Yan H., Gazdar A., Powell D., Riggins G., Willson J., Markowitz S., Kinzler K., Vogelstein B. & Velculescu V.
(2004)
Science.
304,
5670,
p. 554
Bergamaschi D., Samuels Y., Jin B., Duraisingham S., Crook T. & Lu X.
(2004)
Molecular and Cellular Biology.
24,
3,
p. 1341-1350
We recently showed that ASPP1 and ASPP2 stimulate the apoptotic function of p53. We show here that ASPP1 and ASPP2 also induce apoptosis independently of p53. By binding to p63 and p73 in vitro and in vivo, ASPP1 and ASPP2 stimulate the transactivation function of p63 and p73 on the promoters of Bax, PIG3, and PUMA but not mdm2 or p21WAF-1/CIP1. The expression of ASPP1 and ASPP2 also enhances the apoptotic function of p63 and p73 by selectively inducing the expression of endogenous p53 target genes, such as PIG3 and PUMA, but not mdm2 or p21WAF-1/CIP1. Removal of endogenous p63 or p73 with RNA interference demonstrated that (16) the p53-independent apoptotic function of ASPP1 and ASPP2 is mediated mainly by p63 and p73. Hence, ASPP1 and ASPP2 are the first two identified common activators of all p53 family members. All these results suggest that ASPP1 and ASPP2 could suppress tumor growth even in tumors expressing mutant p53.
Bergamaschi D., Samuels Y., O'Neil N. J., Trigiante G., Crook T., Hsieh J. K., O'Connor D. J., Zhong S., Campargue I., Tomlinson M. L., Kuwabara P. E. & Lu X.
(2003)
Nature Genetics.
33,
2,
p. 162-167
We have previously shown that ASPP1 and ASPP2 are specific activators of p53; one mechanism by which wild-type p53 is tolerated in human breast carcinomas is through loss of ASPP activity. We have further shown that 53BP2, which corresponds to a C-terminal fragment of ASPP2, act as a dominant negative inhibitor of p53 (ref. 1). Hence, an inhibitory form of ASPP resembling 53BP2 could allow cells to bypass the tumor-suppressor functions of p53 and the ASPP proteins. Here, we characterize such a protein, iASPP (inhibitory member of the ASPP family), encoded by PPP1R13L in humans and ape-1 in Caenorhabditis elegans. iASPP is an evolutionarily conserved inhibitor of p53; inhibition of iASPP by RNA-mediated interference or antisense RNA in C. elegans or human cells, respectively, induces p53-dependent apoptosis. Moreover, iASPP is an oncoprotein that cooperates with Ras, E1A and E7, but not mutant p53, to transform cells in vitro. Increased expression of iASPP also confers resistance to ultraviolet radiation and to cisplatin-induced apoptosis. iASPP expression is upregulated in human breast carcinomas expressing wild-type p53 and normal levels of ASPP. Inhibition of iASPP could provide an important new strategy for treating tumors expressing wild-type p53.
Almog R., Low M., Cohen D., Robin G., Ashkenazi S., Bercovier H., Gdalevich M., Samuels Y., Ashkenazi I., Shemer J., Eldad A. & Green M. S.
(1999)
Infection.
27,
3,
p. 212-217
The goal of this study was to assess the susceptibility of the sub-population of over 500,000 immigrants from the former USSR who came to Israel during 1989-94 to HAV infection, and to provide military physicians with estimates of the prevalence of HBV and HCV carriage in this sub-population. 987 males aged 17-49 and 195 females aged 17-19, reporting to military recruitment offices between December 1991 and March 1992 were tested. Anti-HAV, anti-HBV antibodies and hepatitis B surface antigen (HBsAg) were detected by using standard enzyme immunoassay (EIA) tests, and anti-HCV antibodies by a second-generation EIA and confirmed by a third-generation INNO-LIA test. It was found that in the 17-19-year age-group the prevalence of anti-HAV antibodies was 37%, anti-HBV was 12.8%, HBsAg was 3.0% and anti-HCV 1.3%. All markers were higher among males. The prevalence of anti-HAV and anti-HBs antibodies increased with age among males. That of HBsAg and anti-HCV antibodies increased with age overall. In the multiple logistic regression analysis, HAV and HBV seropositivity were significantly associated with the mother's education and republic of origin. It was concluded that the prevalence of anti-HAV antibodies is similar to that among the local population, which should not be considered at a higher risk of infection during military service. On the other hand, the higher prevalence of HBsAg and anti-HCV antibodies in this sub-population should heighten the awareness of the possibility of chronic liver pathology.
Avraham A., Jung S., Samuels Y., Seger R. & Ben-Neriah Y.
(1998)
European Journal of Immunology.
28,
8,
p. 2320-2330
Previously we implicated c-Jun N-terminal kinase (JNK) as an element that is involved in signal integration during co-stimulation of T lymphocytes. This pathway has now been traced to an upper level, comprising MAPKK SEK1/MKK4/JNKK1 which, similarly to JNK, must receive input both from the TCR and CD28. A large portion of this input is probably integrated at the level of the Rho-family protein CDC42 which, here, activates SEK1 and JNK to the level reached by TCR and CD28 stimulation. We have identified another putative SEK/JNK pathway regulator, PKCθ, which in contrast to CDC42, activates SEK and JNK maximally only in conjunction with a calcium signal delivered through calcineurin. Signals originating at the TCR and CD28 may travel down the JNK pathway via PKCθ, calcineurin, CDC42, MEKK1 and SEK1.