Kitsberg Y., Nachshon A., Arazi T., Broennimann K., Fisher T., Wainstein A., Finkel Y., Stern-Ginossar N. & Schwartz M.
(2026)
Nature Communications.
17,
1,
1300.
Human cytomegalovirus (HCMV) infection results in either productive or latent infection, the latter enabling life-long viral persistence. Monocytes support latent infection but become permissive to productive infection upon differentiation into macrophages. These differentiation-driven differences have been largely attributed to chromatin-mediated repression of the viral genome. Using metabolic labeling of newly synthesized RNA, we observe markedly lower viral transcription at early stages of infection in monocytes compared to macrophages. Unbiased comparison reveals that this difference is partly explained by inefficient viral entry in monocytes: fewer viruses enter, and correspondingly, fewer genomes reach the nucleus. Indeed, ectopic expression of known HCMV entry receptors in monocytes enhances viral entry and enables productive infection, demonstrating that these cells can support full lytic replication if entry is efficient. We further identify integrin β3 as a differentiation-induced surface protein playing an important role in HCMV entry into macrophages, partially accounting for the observed differences in entry efficiency. Finally, we show that cells receiving fewer viral genomes are the ones that establish latent infection and have the capacity to reactivate. Overall, our findings reveal that entry is a previously unrecognized factor contributing to latent infection in monocytes, adding a critical layer to the paradigm of HCMV latency.
Fisher T., Mizrahi O., Tai-Schmiedel J., Nachshon A., Schwartz M., Patrick M., Gluck A., Aharon E., Karniely S. & Stern-Ginossar N.
(2026)
Molecular Cell.
86,
7,
p. 1247-1259.e8
During infection with human cytomegalovirus (HCMV), the viral long non-coding RNA RNA2.7 becomes the most abundant polyadenylated transcript in the cell, yet its function has remained enigmatic. By combining RNA sequencing, metabolic labeling of newly synthesized RNA, and ribosome profiling, we define how RNA2.7 modulates host gene expression and promotes viral propagation. We show that RNA2.7 stabilizes numerous host mRNAs by sequestering a broad array of RNA-binding proteins, reshaping the cellular transcriptome. Accordingly, RNA2.7 is essential for HCMV-induced cell-cycle arrest at the G1-S transition specifically when infection occurs in G1, thereby enhancing viral replication in actively cycling cells. Notably, RNA2.7 expression alone is sufficient to block cell-cycle progression, and screening RNA2.7 fragments identifies a region containing an extended polyadenosine stretch that is required for this activity. Our findings reveal how RNA2.7 promotes viral replication by modulating host mRNA stability and enforcing cell-cycle arrest, creating favorable conditions for infection.
Aharon-Hefetz N., Schwartz M., Aharon E., Stern-Ginossar N., Dahan O. & Pilpel Y.
(2026)
Molecular Systems Biology.
22,
p. 519-536
Human viruses rely on host translation resources, including the cellular tRNA pool, because they lack tRNA genes. Using tRNA sequencing, we profiled mature tRNAs during infections with human cytomegalovirus (HCMV) and SARS-CoV-2. HCMV-induced alterations in mature tRNA levels were predominantly virus-driven, with minimal influence from the cellular immune response. Certain post-transcriptional modifications, correlated with tRNA stability, were actively manipulated by HCMV. By contrast, SARS-CoV-2 caused minimal changes in mature tRNA levels or modifications. Comparing viral codon usage with proliferation- versus differentiation-associated codon-usage signatures in human genes revealed striking divergence. HCMV genes aligned with differentiation codon usage, whereas SARS-CoV-2 genes matched proliferation codon usage. Structural and gene-expression genes in both viruses showed strong adaptation to host tRNA pools. Finally, a systematic CRISPR screen of human tRNA genes and tRNA-modifying enzymes identified specific tRNAs and enzymes that either enhanced or restricted HCMV infectivity and influenced cellular growth. Together, these data define a dynamic interplay between the host tRNA landscape and viral infection, illuminating the mechanisms governing hostvirus interactions.
Rozman B., Broennimann K., Rajan K. S., Nachshon A., Saha C., Arazi T., Mohan V., Geiger T., Wollner C. J., Richner J. M., Westhof E., Yonath A., Bashan A. & Stern-Ginossar N.
(2026)
Nature.
The considerable success of mRNA vaccines against SARS-CoV-2 has underscored the potential of synthetic mRNA as a transformative biomedical technology1. A critical feature of this approach is the incorporation of the modified nucleoside N1-methylpseudouridine (m1Ψ), which enhances antigen expression while reducing immunogenicity2, 3, 45. However, a comprehensive understanding of how m1Ψ influences translation remains incomplete. Here we use ribosome profiling at the subcodon resolution to show that m1Ψ increases ribosome density on synthetic mRNAs, leading to higher protein production independent of innate immune activation or eIF2α phosphorylation. We find that m1Ψ directly slows ribosome movement in defined sequence contexts while simultaneously promoting translation initiation. Structural studies using cryo-electron microscopy reveal that m1Ψ alters interactions within the ribosomal decoding centre, providing a mechanistic basis for slowed elongation. Furthermore, by introducing synonymous recoding that disrupts the modification-mediated changes in elongation, we show that the m1Ψ-dependent enhancement of protein output is modulated by codon composition, and that m1Ψ impact is strongest in mRNAs containing non-optimal codons with uridines at the wobble position. Together, these findings demonstrate that m1Ψ directly modulates translation dynamics, thereby increasing protein yield from synthetic mRNAs in specific sequence contexts.
CRISPR-Cas9 technology has transformed the study of gene function, enabling the systematic investigation of host-virus interactions. However, most CRISPR-based screens in the context of viral infections rely on cell survival as a readout, which limits their sensitivity and biases results toward early infection stages. To address these challenges, we developed the virus-encoded CRISPR-based direct readout system (VECOS), a virus-centric approach in which human cytomegalovirus is engineered to express single-guide RNA (sgRNA) libraries directly from its genome. This system allows sgRNA abundance, embedded in the viral genome, to serve as a direct and quantitative readout of gene-perturbation effects on viral propagation. By tracking sgRNA levels at distinct stages of the viral infection cycle, VECOS enables a detailed, multidimensional analysis of virus-host interactions. Here we present a modular detailed Protocol for (1) constructing and reconstituting complex sgRNA libraries in double-stranded DNA viruses using bacterial artificial chromosomes, (2) performing multipassage screens to investigate perturbation effects on various stages of viral infection and (3) analyzing the multipassage and multistage sgRNA abundance measurements utilizing a comprehensive framework for data analysis. Successful implementation of this full Protocol takes 14-22 weeks and requires proficiency in molecular biology, as well as basic familiarity with Unix-based computing and programming in R for data processing. This Protocol offers researchers a robust tool for uncovering the molecular mechanisms that drive viral propagation and host-virus interactions.
Finkel Y., Nachshon A., Aharon E., Arazi T., Simonovsky E., Dobešová M., Saud Z., Gluck A., Fisher T., Stanton R. J., Schwartz M. & Stern-Ginossar N.
(2024)
Nature.
630,
8017,
p. 712-719
Genetic screens have transformed our ability to interrogate cellular factor requirements for viral infections1,2, but most current approaches are limited in their sensitivity, biased towards early stages of infection and provide only simplistic phenotypic information that is often based on survival of infected cells24. Here, by engineering human cytomegalovirus to express single guide RNA libraries directly from the viral genome, we developed virus-encoded CRISPR-based direct readout screening (VECOS), a sensitive, versatile, viral-centric approach that enables profiling of different stages of viral infection in a pooled format. Using this approach, we identified hundreds of host dependency and restriction factors and quantified their direct effects on viral genome replication, viral particle secretion and infectiousness of secreted particles, providing a multi-dimensional perspective on virushost interactions. These high-resolution measurements reveal that perturbations altering late stages in the life cycle of human cytomegalovirus (HCMV) mostly regulate viral particle quality rather than quantity, establishing correct virion assembly as a critical stage that is heavily reliant on virushost interactions. Overall, VECOS facilitates systematic high-resolution dissection of the role of human proteins during the infection cycle, providing a roadmap for in-depth study of hostherpesvirus interactions.
Schwartz M. & Stern-Ginossar N.
(2023)
Annals of the New York Academy of Sciences.
1524,
1,
p. 30-36
Human cytomegalovirus (HCMV) is a prevalent herpesvirus, infecting the majority of the human population. Like other herpesviruses, it causes lifelong infection through the establishment of latency. Although reactivation from latency can cause significant morbidity and mortality in immunocompromised hosts, our understanding of HCMV latency and how it is maintained remains limited. Here, we discuss the characterized latency reservoir in hematopoietic cells in the bone marrow and the gaps in our knowledge of mechanisms that facilitate HCMV genome maintenance in dividing cells. We further review clinical evidence that strongly suggests the tissue origin of HCMV reactivation, and we outline similarities to murine cytomegalovirus where latency in tissue-resident cells has been demonstrated. Overall, we think these observations call for a rethinking of HCMV latency reservoirs and point to potential sources of HCMV latency that reside in tissues.
Schwartz M., Shnayder M., Nachshon A., Arazi T., Kitsberg Y., Levi Samia R., Lavi M., Kuint R., Tsabari R. & Stern-Ginossar N.
(2023)
Nature Microbiology.
8,
3,
p. 455-468
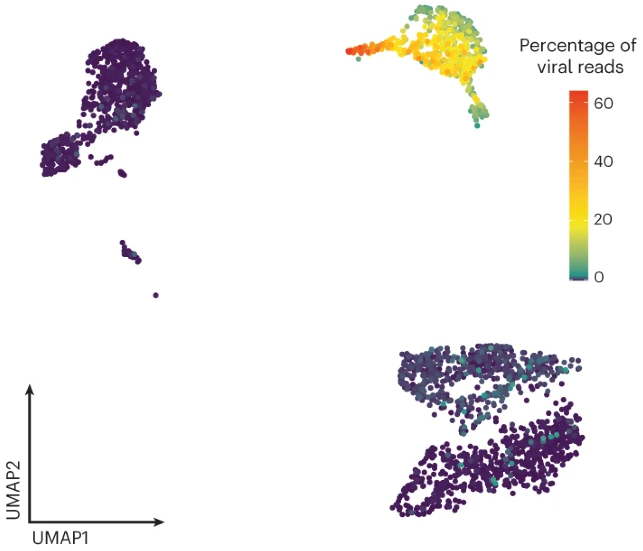
Human cytomegalovirus (HCMV) can result in either productive or non-productive infection, with the latter potentially leading to viral latency. The molecular factors dictating these outcomes are poorly understood. Here we used single-cell transcriptomics to analyse HCMV infection progression in monocytes, which are latently infected, and macrophages, considered to be permissive for productive infection. We show that early viral gene expression levels, specifically of those encoding immediate early proteins IE1 and IE2, are a major factor dictating productive infection. We also revealed that intrinsic, not induced, host cell interferon-stimulated gene expression level is a main determinant of infection outcome. Intrinsic interferon-stimulated gene expression is downregulated with monocyte to macrophage differentiation, partially explaining increased macrophage susceptibility to productive HCMV infection. Furthermore, non-productive macrophages could reactivate, making them potential latent virus reservoirs. Overall, we decipher molecular features underlying HCMV infection outcomes and propose macrophages as a potential HCMV reservoir.
Viral reproduction is contingent on viral protein synthesis that relies on the host ribosomes. As such, viruses have evolved remarkable strategies to hijack the host translational apparatus in order to favor viral protein production and to interfere with cellular innate defenses. Here, we describe the approaches viruses use to exploit the translation machinery, focusing on commonalities across diverse viral families, and discuss the functional relevance of this process. We illustrate the complementary strategies host cells utilize to block viral protein production and consider how cells ensure an efficient antiviral response that relies on translation during this tug of war over the ribosome. Finally, we highlight potential roles mRNA modifications and ribosome quality control play in translational regulation and innate immunity. We address these topics in the context of the COVID-19 pandemic and focus on the gaps in our current knowledge of these mechanisms, specifically in viruses with pandemic potential.
Israeli M., Finkel Y., Yahalom-Ronen Y., Paran N., Chitlaru T., Israeli O., Cohen-Gihon I., Aftalion M., Falach R., Rotem S., Elia U., Nemet I., Kliker L., Mandelboim M., Beth-Din A., Israely T., Cohen O., Stern-Ginossar N. & Bercovich-Kinori A.
(2022)
Nature Communications.
13,
1,
2237.
The global spread of SARS-CoV-2 led to major economic and health challenges worldwide. Revealing host genes essential for infection by multiple variants of SARS-CoV-2 can provide insights into the virus pathogenesis, and facilitate the development of novel therapeutics. Here, employing a genome-scale CRISPR screen, we provide a comprehensive data-set of cellular factors that are exploited by wild type SARS-CoV-2 as well as two additional recently emerged variants of concerns (VOCs), Alpha and Beta. We identified several host factors critical for SARS-CoV-2 infection, including various components belonging to the Clathrin-dependent transport pathway, ubiquitination, Heparan sulfate biogenesis and host phosphatidylglycerol biosynthesis. Comparative analysis of the different VOCs revealed the host factors KREMEN2 and SETDB1 as potential unique candidates required only to the Alpha variant. Furthermore, the analysis identified GATA6, a zinc finger transcription factor, as an essential proviral gene for all variants inspected. We show that GATA6 directly regulates ACE2 transcription and accordingly, is critical for SARS-CoV-2 cell entry. Analysis of clinical samples collected from SARS-CoV-2 infected individuals shows elevated levels of GATA6, suggesting a role in COVID-19 pathogenesis. Finally, pharmacological inhibition of GATA6 resulted in down-modulation of ACE2 and inhibition of viral infectivity. Overall, we show GATA6 may represent a target for the development of anti-SARS-CoV-2 therapeutic strategies and reaffirm the value of the CRISPR loss-of-function screens in providing a list of potential new targets for therapeutic interventions.
David M., Olender T., Mizrahi O., Weingarten-Gabbay S., Friedlander G., Meril S., Goldberg N., Savidor A., Levin Y., Salomon V., Stern-Ginossar N., Bialik S. & Kimchi A.
(2022)
RNA.
28,
10,
p. 1325-1336
Death associated protein 5 (DAP5/eIF4G2/NAT1) is a member of the eIF4G translation initiation factors that has been shown to mediate noncanonical and/or cap-independent translation. It is essential for embryonic development and for differentiation of embryonic stem cells (ESCs), specifically its ability to drive translation of specific target mRNAs. In order to expand the repertoire of DAP5 target mRNAs, we compared ribosome profiles in control and DAP5 knockdown (KD) human ESCs (hESCs) to identify mRNAs with decreased ribosomal occupancy upon DAP5 silencing. A cohort of 68 genes showed decreased translation efficiency in DAP5 KD cells. Mass spectrometry confirmed decreased protein abundance of a significant portion of these targets. Among these was KMT2D, a histone methylase previously shown to be essential for ESC differentiation and embryonic development. We found that nearly half of the cohort of DAP5 target mRNAs displaying reduced translation efficiency of their main coding sequences upon DAP5 KD contained upstream open reading frames (uORFs) that are actively translated independently of DAP5. This is consistent with previously suggested mechanisms by which DAP5 mediates leaky scanning through uORFs and/or reinitiation at the main coding sequence. Crosslinking proteinRNA immunoprecipitation experiments indicated that a significant subset of DAP5 mRNA targets bound DAP5, indicating that direct binding between DAP5 protein and its target mRNAs is a frequent but not absolute requirement for DAP5-dependent translation of the main coding sequence. Thus, we have extended DAP5s function in translation of specific mRNAs in hESCs by a mechanism allowing translation of the main coding sequence following upstream translation of short ORFs.
Fisher T., Gluck A., Narayanan K., Kuroda M., Nachshon A., Hsu J. C., Halfmann P. J., Yahalom-Ronen Y., Tamir H., Finkel Y., Schwartz M., Weiss S., Tseng C. T. K., Israely T., Paran N., Kawaoka Y., Makino S. & Stern-Ginossar N.
(2022)
Cell Reports.
39,
11,
110954.
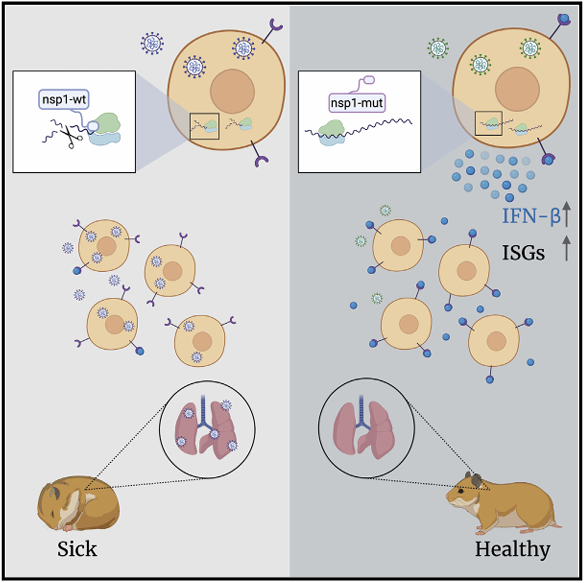
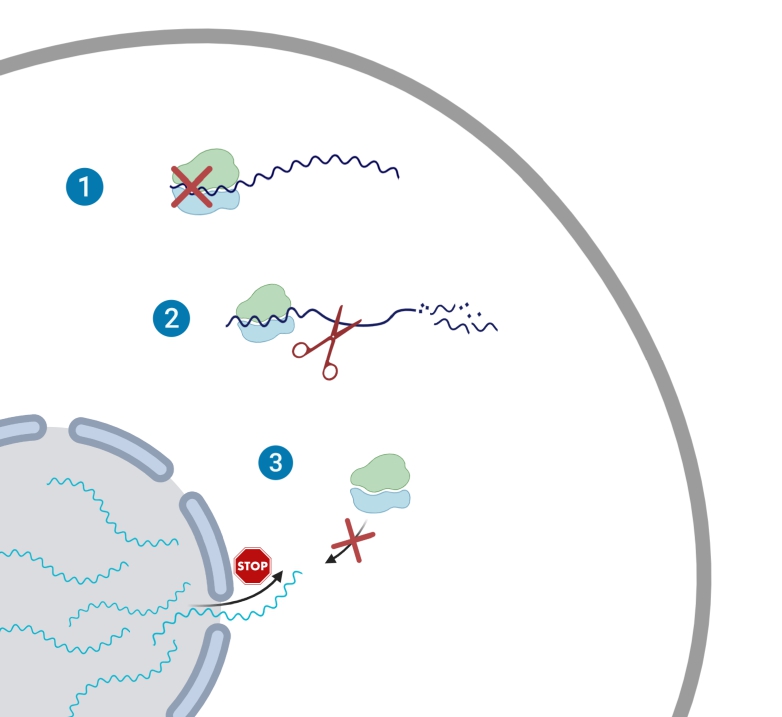
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) leads to shutoff of protein synthesis, and nsp1, a central shutoff factor in coronaviruses, inhibits cellular mRNA translation. However, the diverse molecular mechanisms employed by nsp1 as well as its functional importance are unresolved. By overexpressing various nsp1 mutants and generating a SARS-CoV-2 mutant, we show that nsp1, through inhibition of translation and induction of mRNA degradation, targets translated cellular mRNA and is the main driver of host shutoff during infection. The propagation of nsp1 mutant virus is inhibited exclusively in cells with intact interferon (IFN) pathway as well as in vivo, in hamsters, and this attenuation is associated with stronger induction of type I IFN response. Therefore, although nsp1s shutoff activity is broad, it plays an essential role, specifically in counteracting the IFN response. Overall, our results reveal the multifaceted approach nsp1 uses to shut off cellular protein synthesis and uncover nsp1s explicit role in blocking the IFN response.
Rozman B., Nachshon A., Levi Samia R., Lavi M., Schwartz M. & Stern-Ginossar N.
(2022)
Cell Reports.
39,
2,
110653.
During productive human cytomegalovirus (HCMV) infection, viral genes are expressed in a coordinated cascade that conventionally relies on the dependencies of viral genes on protein synthesis and viral DNA replication. By contrast, the transcriptional landscape of HCMV latency is poorly understood. Here, we examine viral gene expression dynamics during the establishment of both productive and latent HCMV infections. We redefine HCMV gene expression kinetics during productive infection and reveal that viral gene regulation does not represent a simple sequential cascade; many viral genes are regulated by multiple independent modules. Using our improved gene expression classification combined with transcriptome-wide measurements of the effects of a wide array of epigenetic inhibitors on viral gene expression during latency, we show that a defining feature of latency is the unique repression of immediate-early (IE) genes. Altogether, we recharacterize HCMV gene expression kinetics and reveal governing principles of lytic and latent gene expression.
Rak R., Polonsky M., Eizenberg-Magar I., Mo Y., Sakaguchi Y., Mizrahi O., Nachshon A., Reich-Zeliger S., Stern-Ginossar N., Dahan O., Suzuki T., Friedman N. & Pilpel Y.
(2021)
Proceedings of the National Academy of Sciences of the United States of America.
118,
42,
e210655611.
The tRNA pool determines the efficiency, throughput, and accuracy of translation. Previous studies have identified dynamic changes in the tRNA (transfer RNA) supply and mRNA (messenger RNA) demand during cancerous proliferation. Yet dynamic changes may also occur during physiologically normal proliferation, and these are less well characterized. We examined the tRNA and mRNA pools of T cells during their vigorous proliferation and differentiation upon triggering their antigen receptor. We observed a global signature of switch in demand for codons at the early proliferation phase of the response, accompanied by corresponding changes in tRNA expression levels. In the later phase, upon differentiation, the response of the tRNA pool relaxed back to the basal level, potentially restraining excessive proliferation. Sequencing of tRNAs allowed us to evaluate their diverse base-modifications. We found that two types of tRNA modifications, wybutosine and ms2t6A, are reduced dramatically during T cell activation. These modifications occur in the anticodon loops of two tRNAs that decode \u201cslippery codons,\u201d which are prone to ribosomal frameshifting. Attenuation of these frameshift-protective modifications is expected to increase the potential for proteome-wide frameshifting during T cell proliferation. Indeed, human cell lines deleted of a wybutosine writer showed increased ribosomal frameshifting, as detected with an HIV gag-pol frameshifting site reporter. These results may explain HIVs specific tropism toward proliferating T cells since it requires ribosomal frameshift exactly on the corresponding codon for infection. The changes in tRNA expression and modifications uncover a layer of translation regulation during T cell proliferation and expose a potential tradeoff between cellular growth and translation fidelity.
Stern-Ginossar N., Kanneganti T. D., Cameron C. E., Lou Z., Cherry S., Abraham J. & Martin-Sancho L.
(2021)
Molecular Cell.
81,
11,
p. 2261-2265
COVID-19 altered our lives and pushed scientific research to operate at breakneck speed, leading to significant breakthroughs in record time. We asked experts in the field about the challenges they faced in transitioning, rapidly but safely, to working on the virus while navigating the shutdown. Their voices converge on the importance of teamwork, forging new collaborations, and working toward a shared goal.
Uzonyi A., Nir R., Shliefer O., Stern-Ginossar N., Antebi Y., Stelzer Y., Levanon E. Y. & Schwartz S.
(2021)
Molecular Cell.
81,
11,
p. 2374-2387.e3
Adenosine-to-inosine editing is catalyzed by ADAR1 at thousands of sites transcriptome-wide. Despite intense interest in ADAR1 from physiological, bioengineering, and therapeutic perspectives, the rules of ADAR1 substrate selection are poorly understood. Here, we used large-scale systematic probing of ∼2,000 synthetic constructs to explore the structure and sequence context determining editability. We uncover two structural layers determining the formation and propagation of A-to-I editing, independent of sequence. First, editing is robustly induced at fixed intervals of 35 bp upstream and 30 bp downstream of structural disruptions. Second, editing is symmetrically introduced on opposite sites on a double-stranded structure. Our findings suggest a recursive model for RNA editing, whereby the structural alteration induced by the editing at one site iteratively gives rise to the formation of an additional editing site at a fixed periodicity, serving as a basis for the propagation of editing along and across both strands of double-stranded RNA structures.
The coronavirus SARS-CoV-2 is the cause of the ongoing pandemic of COVID-19
1. Coronaviruses have developed a variety of mechanisms to repress host mRNA translation to allow the translation of viral mRNA, and concomitantly block the cellular innate immune response
2,3. Although several different proteins of SARS-CoV-2 have previously been implicated in shutting off host expression
47, a comprehensive picture of the effects of SARS-CoV-2 infection on cellular gene expression is lacking. Here we combine RNA sequencing, ribosome profiling and metabolic labelling of newly synthesized RNA to comprehensively define the mechanisms that are used by SARS-CoV-2 to shut off cellular protein synthesis. We show that infection leads to a global reduction in translation, but that viral transcripts are not preferentially translated. Instead, we find that infection leads to the accelerated degradation of cytosolic cellular mRNAs, which facilitates viral takeover of the mRNA pool in infected cells. We reveal that the translation of transcripts that are induced in response to infection (including innate immune genes) is impaired. We demonstrate this impairment is probably mediated by inhibition of nuclear mRNA export, which prevents newly transcribed cellular mRNA from accessing ribosomes. Overall, our results uncover a multipronged strategy that is used by SARS-CoV-2 to take over the translation machinery and to suppress host defences.
Jungreis I., Nelson C. W., Ardern Z., Finkel Y., Krogan N. J., Sato K., Ziebuhr J., Stern-Ginossar N., Pavesi A., Firth A. E., Gorbalenya A. E. & Kellis M.
(2021)
Virology.
558,
p. 145-151
At least six small alternative-frame open reading frames (ORFs) overlapping well-characterized SARS-CoV-2 genes have been hypothesized to encode accessory proteins. Researchers have used different names for the same ORF or the same name for different ORFs, resulting in erroneous homological and functional inferences. We propose standard names for these ORFs and their shorter isoforms, developed in consultation with the Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. We recommend calling the 39 codon Spike-overlapping ORF ORF2b; the 41, 57, and 22 codon ORF3a-overlapping ORFs ORF3c, ORF3d, and ORF3b; the 33 codon ORF3d isoform ORF3d-2; and the 97 and 73 codon Nucleocapsid-overlapping ORFs ORF9b and ORF9c. Finally, we document conflicting usage of the name ORF3b in 32 studies, and consequent erroneous inferences, stressing the importance of reserving identical names for homologs. We recommend that authors referring to these ORFs provide lengths and coordinates to minimize ambiguity caused by prior usage of alternative names.
Goettsch W., Beerenwinkel N., Deng L., Dölken L., Dutilh B. E., Erhard F., Kaderali L., von Kleist M., Marquet R., Matthijnssens J., McCallin S., McMahon D., Rattei T., Van Rij R. P., Robertson D. L., Schwemmle M., Stern-Ginossar N. & Marz M.
(2021)
Viruses.
13,
5,
766.
Many recent studies highlight the fundamental importance of viruses. Besides their important role as human and animal pathogens, their beneficial, commensal or harmful functions are poorly understood. By developing and applying tailored bioinformatical tools in important virological models, the Marie Skłodowska-Curie Initiative International Training Network VIROINF will provide a better understanding of viruses and the interaction with their hosts. This will open the door to validate methods of improving viral growth, morphogenesis and development, as well as to control strategies against unwanted microorganisms. The key feature of VIROINF is its interdisciplinary nature, which brings together virologists and bioinformaticians to achieve common goals.
Bartok O., Pataskar A., Nagel R., Laos M., Goldfarb E., Hayoun D., Levy R., Körner P., Kreuger I. Z. M., Champagne J., Zaal E. A., Bleijerveld O. B., Huang X., Kenski J., Wargo J., Brandis A., Levin Y., Mizrahi O., Alon M., Lebon S., Yang W., Nielsen M. M., Stern-Ginossar N., Altelaar M., Berkers C. R., Geiger T., Peeper D. S., Olweus J., Samuels Y. & Agami R.
(2021)
Nature.
590,
7845,
p. 332-337
Extensive tumour inflammation, which is reflected by high levels of infiltrating T cells and interferon-γ (IFNγ) signalling, improves the response of patients with melanoma to checkpoint immunotherapy. Many tumours, however, escape by activating cellular pathways that lead to immunosuppression. One such mechanism is the production of tryptophan metabolites along the kynurenine pathway by the enzyme indoleamine 2,3-dioxygenase 1 (IDO1), which is induced by IFNγ. However, clinical trials using inhibition of IDO1 in combination with blockade of the PD1 pathway in patients with melanoma did not improve the efficacy of treatment compared to PD1 pathway blockade alone, pointing to an incomplete understanding of the role of IDO1 and the consequent degradation of tryptophan in mRNA translation and cancer progression. Here we used ribosome profiling in melanoma cells to investigate the effects of prolonged IFNγ treatment on mRNA translation. Notably, we observed accumulations of ribosomes downstream of tryptophan codons, along with their expected stalling at the tryptophan codon. This suggested that ribosomes bypass tryptophan codons in the absence of tryptophan. A detailed examination of these tryptophan-associated accumulations of ribosomes-which we term 'W-bumps'-showed that they were characterized by ribosomal frameshifting events. Consistently, reporter assays combined with proteomic and immunopeptidomic analyses demonstrated the induction of ribosomal frameshifting, and the generation and presentation of aberrant trans-frame peptides at the cell surface after treatment with IFNγ. Priming of naive T cells from healthy donors with aberrant peptides induced peptide-specific T cells. Together, our results suggest that IDO1-mediated depletion of tryptophan, which is induced by IFNγ, has a role in the immune recognition of melanoma cells by contributing to diversification of the peptidome landscape.
Bernshtein B., Nachshon A., Shnayder M., Stern L., Avdic S., Blyth E., Gottlieb D., Abendroth A., Slobedman B., Stern-Ginossar N. & Schwartz M.
(2021)
Frontiers in Cellular and Infection Microbiology.
10,
607470.
Human cytomegalovirus (HCMV) is a widespread pathogen establishing a latent infection in its host. HCMV reactivation is a major health burden in immunocompromised individuals, and is a major cause of morbidity and mortality following hematopoietic stem cell transplantation (HSCT). Here we determined HCMV genomic levels using droplet digital PCR in different peripheral blood mononuclear cell (PBMC) populations in HCMV reactivating HSCT patients. This high sensitivity approach revealed that all PBMC populations harbored extremely low levels of viral DNA at the peak of HCMV DNAemia. Transcriptomic analysis of PBMCs from high-DNAemia samples revealed elevated expression of genes typical of HCMV specific T cells, while regulatory T cell enhancers as well as additional genes related to immune response were downregulated. Viral transcript levels in these samples were extremely low, but remarkably, the detected transcripts were mainly immediate early viral genes. Overall, our data indicate that HCMV DNAemia is associated with distinct signatures of immune response in the blood compartment, however it is not necessarily accompanied by substantial infection of PBMCs and the residual infected PBMCs are not productively infected.
Lasman L., Krupalnik V., Viukov S., Mor N., Aguilera-Castrejon A., Schneir D., Bayerl J., Mizrahi O., Peles S., Tawil S., Sathe S., Nachshon A., Shani T., Zerbib M., Kilimnik I., Aigner S., Shankar A., Mueller J. R., Schwartz S., Stern-Ginossar N., Yeo G. W., Geula S., Novershtern N. & Hanna J. H.
(2020)
Genes and Development.
34,
19-20,
p. 1373-1391
The N6-methyladenosine (m6A) modification is the most prevalent post-transcriptional mRNA modification, regulating mRNA decay and splicing. It plays a major role during normal development, differentiation, and disease progression. The modification is regulated by a set of writer, eraser, and reader proteins. The YTH domain family of proteins consists of three homologous m6A-binding proteins, Ythdf1, Ythdf2, and Ythdf3, which were suggested to have different cellular functions. However, their sequence similarity and their tendency to bind the same targets suggest that they may have overlapping roles. We systematically knocked out (KO) the Mettl3 writer, each of the Ythdf readers, and the three readers together (triple-KO). We then estimated the effect in vivo in mouse gametogenesis, postnatal viability, and in vitro in mouse embryonic stem cells (mESCs). In gametogenesis, Mettl3-KO severity is increased as the deletion occurs earlier in the process, and Ythdf2 has a dominant role that cannot be compensated by Ythdf1 or Ythdf3, due to differences in readers' expression pattern across different cell types, both in quantity and in spatial location. Knocking out the three readers together and systematically testing viable offspring genotypes revealed a redundancy in the readers' role during early development that is Ythdf1/2/3 gene dosage-dependent. Finally, in mESCs there is compensation between the three Ythdf reader proteins, since the resistance to differentiate and the significant effect on mRNA decay occur only in the triple-KO cells and not in the single KOs. Thus, we suggest a new model for the Ythdf readers function, in which there is profound dosage-dependent redundancy when all three readers are equivalently coexpressed in the same cell types.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the cause of the ongoing coronavirus disease 2019 (COVID-19) pandemic 1. To understand the pathogenicity and antigenic potential of SARS-CoV-2 and to develop therapeutic tools, it is essential to profile the full repertoire of its expressed proteins. The current map of SARS-CoV-2 coding capacity is based on computational predictions and relies on homology with other coronaviruses. Since the protein complement varies among coronaviruses, especially in regard to the variety of accessory proteins, it is crucial to characterize the specific range of SARS-CoV-2 proteins in an unbiased and open-ended manner. Using a suite of ribosome-profiling techniques 2-4, we present a high-resolution map of coding regions in the SARS-CoV-2 genome, which enables us to accurately quantify the expression of canonical viral open reading frames (ORFs) and to identify 23 unannotated viral ORFs. These ORFs include upstream ORFs that are likely to have a regulatory role, several in-frame internal ORFs within existing ORFs, resulting in N-terminally truncated products, as well as internal out-of-frame ORFs, which generate novel polypeptides. We further show that viral mRNAs are not translated more efficiently than host mRNAs; instead, virus translation dominates host translation because of the high levels of viral transcripts. Our work provides a resource that will form the basis of future functional studies.
Shulman Z. & Stern-Ginossar N.
(2020)
Nature Immunology.
21,
5,
p. 501-512
The RNA modification N-6-methyladenosine (m(6)A) plays an essential role in the regulation of immunity. Here, Shulman and Stern-Ginossar review the roles of m(6)A in controlling immune recognition, activation of innate and adaptive immune responses, and cell fate decisions.Protection from harmful pathogens depends on activation of the immune system, which relies on tight regulation of gene expression. Recently, the RNA modification N-6-methyladenosine (m(6)A) has been found to play an essential role in such regulation. Here, we summarize newly discovered functions of m(6)A in controlling various aspects of immunity, including immune recognition, activation of innate and adaptive immune responses, and cell fate decisions. We then discuss some of the current challenges in the field and describe future directions for uncovering the immunological functions of m(6)A and its mechanisms of action.
Tai-Schmiedel J., Karniely S., Lau B., Ezra A., Eliyahu E., Nachshon A., Kerr K., Suárez N., Schwartz M., Davison A. J. & Stern-Ginossar N.
(2020)
PLoS Pathogens.
16,
4,
e1008390.
Viruses are known for their extremely compact genomes composed almost entirely of protein-coding genes. Nonetheless, four long noncoding RNAs (lncRNAs) are encoded by human cytomegalovirus (HCMV). Although these RNAs accumulate to high levels during lytic infection, their functions remain largely unknown. Here, we show that HCMV-encoded lncRNA4.9 localizes to the viral nuclear replication compartment, and that its depletion restricts viral DNA replication and viral growth. RNA4.9 is transcribed from the HCMV origin of replication (oriLyt) and forms an RNA-DNA hybrid (R-loop) through its G+C-rich 5 end, which may be important for the initiation of viral DNA replication. Furthermore, targeting the RNA4.9 promoter with CRISPR-Cas9 or genetic relocalization of oriLyt leads to reduced levels of the viral single-stranded DNA-binding protein (ssDBP), suggesting that the levels of ssDBP are coupled to the oriLyt activity. We further identified a similar, oriLyt-embedded, G +C-rich lncRNA in murine cytomegalovirus (MCMV). These results indicate that HCMV RNA4.9 plays an important role in regulating viral DNA replication, that the levels of ssDBP are coupled to the oriLyt activity, and that these regulatory features may be conserved among betaherpesviruses.
Le-trilling V. T. K., Becker T., Nachshon A., Stern-ginossar N., Schöler L., Voigt S., Hengel H. & Trilling M.
(2020)
Cell Reports.
30,
7,
p. 2248-2260.e5
Human cytomegalovirus (HCMV) causes diseases in individuals with immature or compromised immunity. To evade immune control, HCMV evolved numerous antagonists targeting the interferon system at multiple levels. By comparative analysis of naturally arising variants of the most widely studied HCMV strain, AD169, and a panel of targeted mutants, we uncover the UL145 gene as indispensable for STAT2 downregulation. Ribosome profiling confirms the translation of the canonical pUL145 protein (pUL145-Long) and newly identifies a shorter isoform (pUL145-Short). Both isoforms recruit DDB1-containing ubiquitin ligases to induce proteasomal degradation of STAT2. An alanine-scanning mutagenesis discloses the DDB1 interaction motif of pUL145 that resembles the DDB1-binding interface of cellular substrate receptors of DDB1-containing ubiquitin ligases. Thus, pUL145 constitutes a viral DDB1-cullin-associated factor (vDCAF), which mimics cellular DCAFs to exploit the ubiquitin-proteasome system to impede antiviral immunity. Notably, the viral exploitation of the cullins can be targeted to restore the efficacy of the host immune response.
Finkel Y., Schmiedel D., Tai-Schmiedel J., Nachshon A., Winkler R., Dobesova M., Schwartz M., Mandelboim O. & Stern-Ginossar N.
(2020)
eLife.
9,
e50960.
Human herpesvirus-6 (HHV-6) A and B are ubiquitous betaherpesviruses, infecting the majority of the human population. They encompass large genomes and our understanding of their protein coding potential is far from complete. Here, we employ ribosome-profiling and systematic transcript-analysis to experimentally define HHV-6 translation products. We identify hundreds of new open reading frames (ORFs), including upstream ORFs (uORFs) and internal ORFs (iORFs), generating a complete unbiased atlas of HHV-6 proteome. By integrating systematic data from the prototypic betaherpesvirus, human cytomegalovirus, we uncover numerous uORFs and iORFs conserved across betaherpesviruses and we show uORFs are enriched in late viral genes. We identified three highly abundant HHV-6 encoded long non-coding RNAs, one of which generates a non-polyadenylated stable intron appearing to be a conserved feature of betaherpesviruses. Overall, our work reveals the complexity of HHV-6 genomes and highlights novel features conserved between betaherpesviruses, providing a rich resource for future functional studies.
Shnayder M., Nachshon A., Rozman B., Bernstein B., Lavi M., Fein N., Poole E., Avdic S., Blyth E., Gottlieb D., Abendroth A., Slobedman B., Sinclair J., Stern-Ginossar N. & Schwartz M.
(2020)
eLife.
9,
e52168.
Human cytomegalovirus (HCMV) causes a lifelong infection through establishment of latency. Although reactivation from latency can cause life-threatening disease, our molecular understanding of HCMV latency is incomplete. Here we use single cell RNA-seq analysis to characterize latency in monocytes and hematopoietic stem and progenitor cells (HSPCs). In monocytes, we identify host cell surface markers that enable enrichment of latent cells harboring higher viral transcript levels, which can reactivate more efficiently, and are characterized by reduced intrinsic immune response that is important for viral gene expression. Significantly, in latent HSPCs, viral transcripts could be detected only in monocyte progenitors and were also associated with reduced immune-response. Overall, our work indicates that regardless of the developmental stage in which HCMV infects, HCMV drives hematopoietic cells towards a weaker immune-responsive monocyte state and that this anergic-like state is crucial for the virus ability to express its transcripts and to eventually reactivate.
Obiedat A., Charpak-Amikam Y., Tai-Schmiedel J., Seidel E., Mahameed M., Avril T., Stern-Ginossar N., Springuel L., Bolsee J., Gilham D. E., Dipta P., Shmuel M., Chevet E., Mandelboim O. & Tirosh B.
(2019)
Journal of Molecular Medicine.
98,
1,
p. 135-148
The B7 family member, B7H6, is a ligand for the natural killer cell receptor NKp30. B7H6 is hardly expressed on normal tissues, but undergoes upregulation on different types of tumors, implicating it as an attractive target for cancer immunotherapy. The molecular mechanisms that control B7H6 expression are poorly understood. We report that in contrast to other NK cell ligands, endoplasmic reticulum (ER) stress upregulates B7H6 mRNA levels and surface expression. B7H6 induction by ER stress requires protein kinase R-like ER kinase (PERK), one of the three canonical sensors of the unfolded protein response. PERK phosphorylates eIF2 alpha, which regulates protein synthesis and gene expression. Because eIF2 alpha is phosphorylated by several kinases following different stress conditions, the program downstream to eIF2 alpha phosphorylation is called the integrated stress response (ISR). Several drugs were reported to promote the ISR. Nelfinavir and lopinavir, two clinically approved HIV protease inhibitors, promote eIF2 alpha phosphorylation by different mechanisms. We show that nelfinavir and lopinavir sustainably instigate B7H6 expression at their pharmacologically relevant concentrations. As such, ER stress and ISR conditions sensitize melanoma targets to CAR-T cells directed against B7H6. Our study highlights a novel mechanism to induce B7H6 expression and suggests a pharmacological approach to improve B7H6-directed immunotherapy. Key messagesB7H6 is induced by ER stress in a PERK-dependent mechanism. Induction of B7H6 is obtained pharmacologically by HIV protease inhibitors. Exposure of tumor cells to the HIV protease inhibitor nelfinavir improves the recognition by B7H6-directed CAR-T.
Poole E., Huang C. J., Forbester J., Shnayder M., Nachshon A., Kweider B., Basaj A., Smith D., Jackson S. E., Liu B., Shih J., Kiskin F. N., Roche K., Murphy E., Wills M. R., Morrell N. W., Dougan G., Stern-Ginossar N., Rana A. A. & Sinclair J.
(2019)
Frontiers in Microbiology.
10,
2233.
Herpesviruses undergo life-long latent infection which can be life-threatening in the immunocompromised. Models of latency and reactivation of human cytomegalovirus (HCMV) include primary myeloid cells, cells known to be important for HCMV latent carriage and reactivation in vivo. However, primary cells are limited in availability, and difficult to culture and to genetically modify; all of which have hampered our ability to fully understand virus/host interactions of this persistent human pathogen. We have now used iPSCs to develop a model cell system to study HCMV latency and reactivation in different cell types after their differentiation down the myeloid lineage. Our results show that iPSCs can effectively mimic HCMV latency/reactivation in primary myeloid cells, allowing molecular interrogations of the viral latent/lytic switch. This model may also be suitable for analysis of other viruses, such as HIV and Zika, which also infect cells of the myeloid lineage.
Rho-Associated Coiled-Coil Kinase 1 Translocates to the Nucleus and Inhibits Human Cytomegalovirus Propagation
Eliyahu E., Tirosh O., Dobesova M., Nachshon A., Schwartz M. & Stern-Ginossar N.
(2019)
Journal of Virology.
93,
19,
00453-19.
Rho-associated coiled-coil kinase (ROCK) protein is a central kinase that regulates numerous cellular functions, including cellular polarity, motility, proliferation, and apoptosis. Here, we demonstrate that ROCK has antiviral properties, and inhibition of its activity results in enhanced propagation of human cytomegalovirus (HCMV). We show that during HCMV infection, ROCK1 translocates to the nucleus and concentrates in the nucleolus, where it colocalizes with the stress-related chaperone heat shock cognate 71-kDa protein (Hsc70). Gene expression measurements show that inhibition of ROCK activity does not seem to affect the cellular stress response. We demonstrate that inhibition of myosin, one of the central targets of ROCK, also increases HCMV propagation, implying that the antiviral activity of ROCK might be mediated by activation of the actomyosin network. Finally, we demonstrate that inhibition of ROCK results in increased levels of the tegument protein UL32 and of viral DNA in the cytoplasm, suggesting ROCK activity hinders the efficient egress of HCMV particles out of the nucleus. Altogether, our findings illustrate ROCK activity restricts HCMV propagation and suggest this inhibitory effect may be mediated by suppression of capsid egress out of the nucleus.IMPORTANCE ROCK is a central kinase in cells that regulates numerous cellular functions, including cellular polarity, motility, proliferation, and apoptosis. Here we reveal a novel antiviral activity of ROCK during infection with HCMV, a prevalent pathogen infecting most of the population worldwide. We reveal ROCK1 is translocated to the nucleus, where it mainly localizes to the nucleolus. Our findings suggest that ROCK's antiviral activity may be related to activation of the actomyosin network and inhibition of capsid egress out of the nucleus.
Malakar P., Stein I., Saragovi A., Winkler R., Stern-Ginossar N., Berger M., Pikarsky E. & Karni R.
(2019)
Cancer Research.
79,
10,
p. 2480-2493
Reprogrammed glucose metabolism of enhanced aerobic glycolysis (or the Warburg effect) is known as a hallmark of cancer. The roles of long noncoding RNAs (lncRNA) in regulating cancer metabolism at the level of both glycolysis and gluconeogenesis are mostly unknown. We previously showed that lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) acts as a proto-oncogene in hepatocellular carcinoma (HCC). Here, we investigated the role of MALAT1 in regulating cancer glucose metabolism. MALAT1 upregulated the expression of glycolytic genes and downregulated gluconeogenic enzymes by enhancing the translation of the metabolic transcription factor TCF7L2. MALAT1-enhanced TCF7L2 translation was mediated by upregulation of SRSF1 and activation of the mTORC1-4EBP1 axis. Pharmacological or genetic inhibition of mTOR and Raptor or expression of a hypophosphorylated mutant version of eIF4E-binding protein (4EBP1) resulted in decreased expression of TCF7L2. MALAT1 expression regulated TCF7L2 mRNA association with heavy polysomes, probably through the TCF7L2 5'-untranslated region (UTR), as determined by polysome fractionation and 5'UTR-reporter assays. Knockdown of TCF7L2 in MALAT1-overexpressing cells and HCC cell lines affected their metabolism and abolished their tumorigenic potential, suggesting that the effects of MALAT1 on glucose metabolism are essential for its oncogenic activity. Taken together, our findings suggest that MALAT1 contributes to HCC development and tumor progression by reprogramming tumor glucose metabolism.
Stern-Ginossar N., Thompson S. R., Mathews M. B. & Mohr I.
(2019)
Cold Spring Harbor perspectives in biology.
11,
3,
a033001.
As obligate intracellular parasites, virus reproduction requires host cell functions. Despite variations in genome size and configuration, nucleic acid composition, and their repertoire of encoded functions, all viruses remain unconditionally dependent on the protein synthesis machinery resident within their cellular hosts to translate viral messenger RNAs (mRNAs). A complex signaling network responsive to physiological stress, including infection, regulates host translation factors and ribosome availability. Furthermore, access to the translation apparatus is patrolled by powerful host immune defenses programmed to restrict viral invaders. Here, we review the tactics and mechanisms used by viruses to appropriate control over host ribosomes, subvert host defenses, and dominate the infected cell translational landscape. These not only define aspects of infection biology paramount for virus reproduction, but continue to drive fundamental discoveries into how cellular protein synthesis is controlled in health and disease.
Winkler R., Gillis E., Lasman L., Safra M., Geula S., Soyris C., Nachshon A., Tai-Schmiedel J., Friedman N., Le-Trilling V. T. K., Trilling M., Mandelboim M., Hanna J. H., Schwartz S. & Stern-Ginossar N.
(2019)
Nature Immunology.
20,
2,
p. 173-182
N6-methyladenosine (m6A) is the most common mRNA modification. Recent studies have revealed that depletion of m6A machinery leads to alterations in the propagation of diverse viruses. These effects were proposed to be mediated through dysregulated methylation of viral RNA. Here we show that following viral infection or stimulation of cells with an inactivated virus, deletion of the m6A writer METTL3 or reader YTHDF2 led to an increase in the induction of interferon-stimulated genes. Consequently, propagation of different viruses was suppressed in an interferon-signaling-dependent manner. Significantly, the mRNA of IFNB, the gene encoding the main cytokine that drives the type I interferon response, was m6A modified and was stabilized following repression of METTL3 or YTHDF2. Furthermore, we show that m6A-mediated regulation of interferon cxgenes was conserved in mice. Together, our findings uncover the role m6A serves as a negative regulator of interferon response by dictating the fast turnover of interferon mRNAs and consequently facilitating viral propagation.
Schwartz M. & Stern-Ginossar N.
(2019)
Journal of Virology.
93,
11,
e0004719.
The latent human cytomegalovirus (HCMV) transcriptome has been extremely difficult to define due to the scarcity of naturally latent cells and the complexity of available models. The genomic era offers many approaches to transcriptome profiling that hold great potential for elucidating this challenging issue. The results from two recent studies applying different transcriptomic methodologies and analyses of both experimental and natural samples challenge the dogma of a restricted latency-associated transcription program. Instead, they portray the hallmark of HCMV latent infection as low-level expression of a broad spectrum of canonical viral lytic genes.
mRNAs carry two layers of information, the genetic code and the information that dictates their post-transcriptional fate. The latter function relies on a complex interplay between cis-elements and trans-regulators, and unbiased identification of these elements is still challenging. To identify cis-elements that control gene expression, we use dimethyl sulfate (DMS) mutational profiling with sequencing and map changes in mRNA secondary structure following viral infection. Our dynamic structural data reveal a major role for ribosomes in unwinding secondary structures, which is further supported by the relationship we uncover between structure and translation efficiency. Moreover, our analysis revealed dozens of regions in viral and cellular mRNAs that exhibit changes in secondary structure. In-depth analysis of these regions reveals cis-elements in 3' UTRs that regulate mRNA stability and elements within coding sequences that control translation. Overall, our study demonstrates how mapping dynamic changes in mRNA structure allows unbiased identification of functional regulatory elements.
Lee J. S., Adler L., Karathia H., Carmel N., Rabinovich S., Auslander N., Keshet R., Stettner N., Silberman A., Agemy L., Helbling D., Eilam R., Sun Q., Brandis A., Malitsky S., Itkin M., Weiss H., Pinto S., Kalaora S., Levy R., Barnea E., Admon A., Dimmock D., Stern-Ginossar N., Scherz A., Nagamani S. C., Unda M., Wilson D. M., Elhasid R., Carracedo A., Samuels Y., Hannenhalli S., Ruppin E. & Erez A.
(2018)
Cell.
174,
6,
p. 1559-1570.e22
The urea cycle (UC) is the main pathway by which mammals dispose of waste nitrogen. We find that specific alterations in the expression of most UC enzymes occur in many tumors, leading to a general metabolic hallmark termed \u201cUC dysregulation\u201d (UCD). UCD elicits nitrogen diversion toward carbamoyl-phosphate synthetase2, aspartate transcarbamylase, and dihydrooratase (CAD) activation and enhances pyrimidine synthesis, resulting in detectable changes in nitrogen metabolites in both patient tumors and their bio-fluids. The accompanying excess of pyrimidine versus purine nucleotides results in a genomic signature consisting of transversion mutations at the DNA, RNA, and protein levels. This mutational bias is associated with increased numbers of hydrophobic tumor antigens and a better response to immune checkpoint inhibitors independent of mutational load. Taken together, our findings demonstrate that UCD is a common feature of tumors that profoundly affects carcinogenesis, mutagenesis, and immunotherapy response. Urea cycle dysregulation (UCD) in cancer is a prevalent phenomenon in multiple cancers. UCD increases nitrogen utilization for pyrimidine synthesis, generating nucleotide imbalance that leads to detectable mutation patterns and biochemical signatures in cancer patients samples. UCD is associated with a worse prognosis but a better response to immunotherapy.
Finkel Y., Stern-Ginossar N. & Schwartz M.
(2018)
Proteomics.
18,
10,
1700255.
Definition of functional genomic elements is one of the greater challenges of the genomic era. Traditionally, putative short open reading frames (sORFs) coding for less than 100 amino acids were disregarded due to computational and experimental limitations; however, it has become clear over the past several years that translation of sORFs is pervasive and serves diverse functions. The development of ribosome profiling, allowing identification of translated sequences genome wide, revealed wide spread, previously unidentified translation events. New computational methodologies as well as improved mass spectrometry approaches also contributed to the task of annotating translated sORFs in different organisms. Viruses are of special interest due to the selective pressure on their genome size, their rapid and confining evolution, and the potential contribution of novel peptides to the host immune response. Indeed, many functional viral sORFs were characterized to date, and ribosome profiling analyses suggest that this may be the tip of the iceberg. Our computational analyses of sORFs identified by ribosome profiling in DNA viruses demonstrate that they may be enriched in specific features implying that at least some of them are functional. Combination of systematic genome editing strategies with synthetic tagging will take us into the next stepelucidation of the biological relevance and function of this intriguing class of molecules.
Toledano T., Vitenshtein A., Stern-Ginossar N., Seidel E. & Mandelboim O.
(2018)
Journal of Immunology.
200,
8,
p. 2819-2825
Recognition of the human stress-induced ligand MHC class I polypeptide-related sequence A (MICA) by the receptor NKG2D expressed on NK cells leads to NK cell mediated killing of the target cells. Hence, the expression of MICA must be tightly regulated, and its cell surface expression needs to be quickly downregulated to avoid inappropriate activation of immune cells. In this article, we describe a transcript variant of human MICA that has not yet been studied, which contains a 3' untranslated region of 119 nt instead of 174. We identify its polyadenylation signal and demonstrate that, upon stresses, such as heat shock, butyrate treatment, and some oxidative and DNA-damaging treatments, the balance between the two MICA variants changes in favor of the less stable, longer variant. Mechanistically, we showed that this change is linked to microRNA activity and that poly ADP ribose polymerase 1 is involved in the induction of the longer variant following stress. Thus, to our knowledge, we identify the first regulatory mechanism of a stress ligand's decay and also provide one of the first physiological examples for the biological function of a longer 3' untranslated region of a particular gene.
Shnayder M., Nachshon A., Krishna B., Poole E., Boshkov A., Binyamin A., Maza I., Sinclair J., Schwartz M. & Stern-Ginossar N.
(2018)
mBio.
9,
2,
e00013-18.
Primary infection with human cytomegalovirus (HCMV) results in a lifelong infection due to its ability to establish latent infection, with one characterized viral reservoir being hematopoietic cells. Although reactivation from latency causes serious disease in immunocompromised individuals, our molecular understanding of latency is limited. Here, we delineate viral gene expression during natural HCMV persistent infection by analyzing the massive transcriptome RNA sequencing (RNA-seq) atlas generated by the Genotype-Tissue Expression (GTEx) project. This systematic analysis reveals that HCMV persistence in vivo is prevalent in diverse tissues. Notably, we find only viral transcripts that resemble gene expression during various stages of lytic infection with no evidence of any highly restricted latency-associated viral gene expression program. To further define the transcriptional landscape during HCMV latent infection, we also used single-cell RNA-seq and a tractable experimental latency model. In contrast to some current views on latency, we also find no evidence for any highly restricted latency-associated viral gene expression program. Instead, we reveal that latency-associated gene expression largely mirrors a late lytic viral program, albeit at much lower levels of expression. Overall, our work has the potential to revolutionize our understanding of HCMV persistence and suggests that latency is governed mainly by quantitative changes, with a limited number of qualitative changes, in viral gene expression. IMPORTANCE Human cytomegalovirus is a prevalent pathogen, infecting most of the population worldwide and establishing lifelong latency in its hosts. Although reactivation from latency causes significant morbidity and mortality in immunocompromised hosts, our molecular understanding of the latent state remains limited. Here, we examine the viral gene expression during natural and experimental latent HCMV infection on a transcriptome-wide level. In contrast to the classical views on herpesvirus latency, we find no evidence for a restricted latency-associated viral gene expression program. Instead, we reveal that latency gene expression largely resembles a late lytic viral profile, albeit at much lower levels of expression. Taken together, our data transform the current view of HCMV persistence and suggest that latency is mainly governed by quantitative rather than qualitative changes in viral gene expression.
Jafarnejad S. M., Chapat C., Matta-Camacho E., Gelbart I. A., Hesketh G. G., Arguello M., Garzia A., Kim S. H., Attig J., Shapiro M., Morita M., Khoutorsky A., Alain T., Christos G. G., Stern-Ginossar N., Tuschl T., Gingras A. C., Duchaine T. F. & Sonenberg N.
(2018)
eLife.
7,
e35034.
MicroRNAs (miRNAs) exert a broad influence over gene expression by directing effector activities that impinge on translation and stability of mRNAs. We recently discovered that the cap-binding protein 4EHP is a key component of the mammalian miRNA-Induced Silencing Complex (miRISC), which mediates gene silencing. However, little is known about the mRNA repertoire that is controlled by the 4EHP/miRNA mechanism or its biological importance. Here, using ribosome profiling, we identify a subset of mRNAs that are translationally controlled by 4EHP. We show that the Dusp6 mRNA, which encodes an ERK1/2 phosphatase, is translationally repressed by 4EHP and a specific miRNA, miR-145. This promotes ERK1/2 phosphorylation, resulting in augmented cell growth and reduced apoptosis. Our findings thus empirically define the integral role of translational repression in miRNA-induced gene silencing and reveal a critical function for this process in the control of the ERK signaling cascade in mammalian cells.
Solomon O., Di Segni A., Cesarkas K., Porath H. T., Marcu-Malina V., Mizrahi O., Stern-Ginossar N., Kol N., Farage-Barhom S., Glick-Saar E., Lerenthal Y., Levanon E. Y., Amariglio N., Unger R., Goldstein I., Eyal E. & Rechavi G.
(2017)
Nature Communications.
8,
1,
1440.
Adenosine deaminase acting on RNA 1 (ADAR1) is the master RNA editor, catalyzing the deamination of adenosine to inosine. RNA editing is vital for preventing abnormal activation of cytosolic nucleic acid sensing pathways by self-double-stranded RNAs. Here we determine, by parallel analysis of RNA secondary structure sequencing (PARS-seq), the global RNA secondary structure changes in ADAR1 deficient cells. Surprisingly, ADAR1 silencing resulted in a lower global double-stranded to single-stranded RNA ratio, suggesting that A-to-I editing can stabilize a large subset of imperfect RNA duplexes. The duplexes destabilized by editing are composed of vastly complementary inverted Alus found in untranslated regions of genes performing vital biological processes, including housekeeping functions and type-I interferon responses. They are predominantly cytoplasmic and generally demonstrate higher ribosomal occupancy. Our findings imply that the editing effect on RNA secondary structure is context dependent and underline the intricate regulatory role of ADAR1 on global RNA secondary structure.
Safra M., Sas-Chen A., Nir R., Winkler R., Nachshon A., Bar-Yaacov D., Erlacher M., Rossmanith W., Stern-Ginossar N. & Schwartz S.
(2017)
Nature.
551,
7679,
p. 251-255
Modifications on mRNA offer the potential of regulating mRNA fate post-transcriptionally. Recent studies suggested the widespread presence of N 1 -methyladenosine (m 1 A), which disrupts Watson-Crick base pairing, at internal sites of mRNAs. These studies lacked the resolution of identifying individual modified bases, and did not identify specific sequence motifs undergoing the modification or an enzymatic machinery catalysing them, rendering it challenging to validate and functionally characterize putative sites. Here we develop an approach that allows the transcriptome-wide mapping of m 1 A at single-nucleotide resolution. Within the cytosol, m 1 A is present in a low number of mRNAs, typically at low stoichiometries, and almost invariably in tRNA T-loop-like structures, where it is introduced by the TRMT6/TRMT61A complex. We identify a single m 1 A site in the mitochondrial ND5 mRNA, catalysed by TRMT10C, with methylation levels that are highly tissue specific and tightly developmentally controlled. m 1 A leads to translational repression, probably through a mechanism involving ribosomal scanning or translation. Our findings suggest that m 1 A on mRNA, probably because of its disruptive impact on base pairing, leads to translational repression, and is generally avoided by cells, while revealing one case in mitochondria where tight spatiotemporal control over m 1 A levels was adopted as a potential means of post-transcriptional regulation.
Moor A. E., Golan M., Massasa E. E., Lemze D., Weizman T., Shenhav R., Baydatch S., Mizrahi O., Winkler R., Golani O., Stern-Ginossar N. & Itzkovitz S.
(2017)
Science.
357,
6357,
p. 1299-1303
Asymmetric messenger RNA (mRNA) localization facilitates efficient translation in cells such as neurons and fibroblasts. However, the extent and importance of mRNA polarization in epithelial tissues are unclear. Here, we used single-molecule transcript imaging and subcellular transcriptomics to uncover global apical-basal intracellular polarization of mRNA in the mouse intestinal epithelium. The localization of mRNAs did not generally overlap protein localization. Instead, ribosomes were more abundant on the apical sides, and apical transcripts were consequently more efficiently translated. Refeeding of fasted mice elicited a basal-to-apical shift in polarization of mRNAs encoding ribosomal proteins, which was associated with a specific boost in their translation. This led to increased protein production, required for efficient nutrient absorption. These findings reveal a posttranscriptional regulatory mechanism involving dynamic polarization of mRNA and polarized translation.
Chapat C., Jafarnejad S. M., Matta-Camacho E., Hesketh G. G., Gelbart I. A., Attig J., Gkogkas C. G., Alain T., Stern-Ginossar N., Fabian M. R., Gingras A., Duchaine T. F. & Sonenberg N.
(2017)
Proceedings of the National Academy of Sciences of the United States of America.
114,
21,
p. 5425-5430
MicroRNAs (miRNAs) play critical roles in a broad variety of biological processes by inhibiting translation initiation and by destabilizing target mRNAs. The CCR4-NOT complex effects miRNA-mediated silencing, at least in part through interactions with 4E-T (eIF4E transporter) protein, but the precise mechanism is unknown. Here we show that the cap-binding eIF4E-homologous protein 4EHP is an integral component of the miRNA-mediated silencing machinery. We demonstrate that the cap-binding activity of 4EHP contributes to the translational silencing by miRNAs through the CCR4-NOT complex. Our results show that 4EHP competes with eIF4E for binding to 4E-T, and this interaction increases the affinity of 4EHP for the cap. We propose a model wherein the 4E-T/4EHP interaction engenders a closed-loopmRNA conformation that blocks translational initiation of miRNA targets.
Bercovich-Kinori A., Tai J., Gelbart I. A., Shitrit A., Ben-Moshe S., Drori Y., Itzkovitz S., Mandelboim M. & Stern-Ginossar N.
(2016)
eLife.
5,
AUGUST,
e18311.
Host shutoff is a common strategy used by viruses to repress cellular mRNA translation and concomitantly allow the efficient translation of viral mRNAs. Here we use RNA-sequencing and ribosome profiling to explore the mechanisms that are being utilized by the Influenza A virus (IAV) to induce host shutoff. We show that viral transcripts are not preferentially translated and instead the decline in cellular protein synthesis is mediated by viral takeover on the mRNA pool. Our measurements also uncover strong variability in the levels of cellular transcripts reduction, revealing that short transcripts are less affected by IAV. Interestingly, these mRNAs that are refractory to IAV infection are enriched in cell maintenance processes such as oxidative phosphorylation. Furthermore, we show that the continuous oxidative phosphorylation activity is important for viral propagation. Our results advance our understanding of IAV-induced shutoff, and suggest a mechanism that facilitates the translation of genes with important housekeeping functions.
Tichon A., Gil N., Lubelsky Y., Solomon T. H., Lemze D., Itzkovitz S., Stern-Ginossar N. & Ulitsky I.
(2016)
Nature Communications.
7,
12209.
Thousands of long noncoding RNA (lncRNA) genes are encoded in the human genome, and hundreds of them are evolutionarily conserved, but their functions and modes of action remain largely obscure. Particularly enigmatic lncRNAs are those that are exported to the cytoplasm, including NORAD - an abundant and highly conserved cytoplasmic lncRNA. Here we show that most of the sequence of NORAD is comprised of repetitive units that together contain at least 17 functional binding sites for the two mammalian Pumilio homologues. Through binding to PUM1 and PUM2, NORAD modulates the mRNA levels of their targets, which are enriched for genes involved in chromosome segregation during cell division. Our results suggest that some cytoplasmic lncRNAs function by modulating the activities of RNA-binding proteins, an activity which positions them at key junctions of cellular signalling pathways.
Vitenshtein A., Weisblum Y., Hauka S., Halenius A., Oiknine-Djian E., Tsukerman P., Bauman Y., Bar-On Y., Stern-Ginossar N., Enk J., Ortenberg R., Tai J., Markel G., Blumberg R. S., Hengel H., Jonjic S., Wolf D. G., Adler H., Kammerer R. & Mandelboim O.
(2016)
Cell Reports.
15,
11,
p. 2331-2339
Cells in our body can induce hundreds of antiviral genes following virus sensing, many of which remain largely uncharacterized. CEACAM1 has been previously shown to be induced by various innate systems; however, the reason for such tight integration to innate sensing systems was not apparent. Here, we show that CEACAM1 is induced following detection of HCMV and influenza viruses by their respective DNA and RNA innate sensors, IFI16 and RIG-I. This induction is mediated by IRF3, which bound to an ISRE element present in the human, but not mouse, CEACAM1 promoter. Furthermore, we demonstrate that, upon induction, CEACAM1 suppresses both HCMV and influenza viruses in an SHP2-dependent process and achieves this broad antiviral efficacy by suppressing mTOR-mediated protein biosynthesis. Finally, we show that CEACAM1 also inhibits viral spread in ex vivo human decidua organ culture.
Dar D., Shamir M., Mellin J. R., Koutero M., Stern-Ginossar N., Cossart P. & Sorek R.
(2016)
Science.
352,
6282,
aad9822.
Riboswitches and attenuators are cis-regulatory RNA elements, most of which control bacterial gene expression via metabolite-mediated, premature transcription termination. We developed an unbiased experimental approach for genome-wide discovery of such ribo-regulators in bacteria. We also devised an experimental platform that quantitatively measures the in vivo activity of all such regulators in parallel and enables rapid screening for ribo-regulators that respond to metabolites of choice. Using this approach, we detected numerous antibiotic-responsive ribo-regulators that control antibiotic resistance genes in pathogens and in the human microbiome. Studying one such regulator in Listeria monocytogenes revealed an attenuation mechanism mediated by antibiotic-stalled ribosomes. Our results expose broad roles for conditional termination in regulating antibiotic resistance and provide a tool for discovering riboswitches and attenuators that respond to previously unknown ligands.
Weingarten-Gabbay S., Elias-Kirma S., Nir R., Gritsenko A. A., Stern-Ginossar N., Yakhini Z., Weinberger A. & Segal E.
(2016)
Science.
351,
6270,
aad4939.
To investigate gene specificity at the level of translation in both the human genome and viruses, we devised a high-throughput bicistronic assay to quantify cap-independent translation.We uncovered thousands of novel cap-independent translation sequences, and we provide insights on the landscape of translational regulation in both humans and viruses.We find extensive translational elements in the 3? untranslated region of human transcripts and the polyprotein region of uncapped RNA viruses. Through the characterization of regulatory elements underlying cap-independent translation activity, we identify potential mechanisms of secondary structure, short sequence motif, and base pairing with the 18S ribosomal RNA (rRNA). Furthermore, we systematically map the 18S rRNA regions for which reverse complementarity enhances translation. Thus, we make available insights into the mechanisms of translational control in humans and viruses.
Fields A. P., Rodriguez E. H., Jovanovic M., Stern-Ginossar N., Haas B. J., Mertins P., Raychowdhury R., Hacohen N., Carr S. A., Ingolia N. T., Regev A. & Weissman J. S.
(2015)
Molecular Cell.
60,
5,
p. 816-827
A fundamental goal of genomics is to identify the complete set of expressed proteins. Automated annotation strategies rely on assumptions about protein-coding sequences (CDSs), e.g., they are conserved, do not overlap, and exceed a minimum length. However, an increasing number of newly discovered proteins violate these rules. Here we present an experimental and analytical framework, based on ribosome profiling and linear regression, for systematic identification and quantification of translation. Application of this approach to lipopolysaccharide-stimulated mouse dendritic cells and HCMV-infected human fibroblasts identifies thousands of novel CDSs, including micropeptides and variants of known proteins, that bear the hallmarks of canonical translation and exhibit translation levels and dynamics comparable to that of annotated CDSs. Remarkably, many translation events are identified in both mouse and human cells even when the peptide sequence is not conserved. Our work thus reveals an unexpected complexity to mammalian translation suited to provide both conserved regulatory or protein-based functions. Fields et al. describe a ribosome profiling-based approach for empirical annotation of protein-coding regions of the genome. Of the thousands of previously unknown translated ORFs they identify in mouse and human, many are conserved or dynamically regulated. Surprisingly, a considerable subset is translated in both species despite weak sequence conservation.
Tirosh O., Cohen Y., Shitrit A., Shani O., Le-Trilling V. T. K., Trilling M., Friedlander G., Tanenbaum M. & Stern-Ginossar N.
(2015)
PLoS Pathogens.
11,
11,
e1005288.
Viruses are by definition fully dependent on the cellular translation machinery, and develop diverse mechanisms to co-opt this machinery for their own benefit. Unlike many viruses, human cytomegalovirus (HCMV) does suppress the host translation machinery, and the extent to which translation machinery contributes to the overall pattern of viral replication and pathogenesis remains elusive. Here, we combine RNA sequencing and ribosomal profiling analyses to systematically address this question. By simultaneously examining the changes in transcription and translation along HCMV infection, we uncover extensive transcriptional control that dominates the response to infection, but also diverse and dynamic translational regulation for subsets of host genes. We were also able to show that, at late time points in infection, translation of viral mRNAs is higher than that of cellular mRNAs. Lastly, integration of our translation measurements with recent measurements of protein abundance enabled comprehensive identification of dozens of host proteins that are targeted for degradation during HCMV infection. Since targeted degradation indicates a strong biological importance, this approach should be applicable for discovering central host functions during viral infection. Our work provides a framework for studying the contribution of transcription, translation and degradation during infection with any virus.
Tanenbaum M. E., Stern-Ginossar N., Weissman J. S. & Vale R. D.
(2015)
eLife.
4,
AUGUST2015,
e07957.
Passage through mitosis is driven by precisely-timed changes in transcriptional regulation and protein degradation. However, the importance of translational regulation during mitosis remains poorly understood. Here, using ribosome profiling, we find both a global translational repression and identified ∼200 mRNAs that undergo specific translational regulation at mitotic entry. In contrast, few changes in mRNA abundance are observed, indicating that regulation of translation is the primary mechanism of modulating protein expression during mitosis. Interestingly, 91% of the mRNAs that undergo gene-specific regulation in mitosis are translationally repressed, rather than activated. One of the most pronounced translationally-repressed genes is Emi1, an inhibitor of the anaphase promoting complex (APC) which is degraded during mitosis. We show that full APC activation requires translational repression of Emi1 in addition to its degradation. These results identify gene-specific translational repression as a means of controlling the mitotic proteome, which may complement post-translational mechanisms for inactivating protein function.
Geula S., Moshitch-Moshkovitz S., Dominissini D., Mansour A. A., Kol N., Salmon-Divon M., Hershkovitz V., Peer E., Mor N., Manor Y. S., Ben-Haim M., Eyal E., Yunger S., Pinto Y., Jaitin D. A., Viukov S., Rais Y., Krupalnik V., Chomsky E., Zerbib M., Maza I., Rechavi Y., Massarwa R., Hanna S., Amit I., Levanon E. Y., Amariglio N., Stern-Ginossar N., Novershtern N., Rechavi G. & Hanna J. H.
(2015)
Science.
347,
6225,
p. 1002-1006
Naïve and primed pluripotent states retain distinct molecular properties, yet limited knowledge exists on how their state transitions are regulated. Here, we identify Mettl3, an N6-methyladenosine (m6A) transferase, as a regulator for terminating murine naïve pluripotency. Mettl3 knockout preimplantation epiblasts and naïve embryonic stem cells are depleted for m6A inmRNAs, yet are viable. However, they fail to adequately terminate their naïve state and, subsequently, undergo aberrant and restricted lineage priming at the postimplantation stage, which leads to early embryonic lethality. m6A predominantly and directly reduces mRNA stability, including that of key naïve pluripotency-promoting transcripts. This study highlights a critical role for an mRNA epigenetic modification in vivo and identifies regulatory modules that functionally influence naïve and primed pluripotency in an opposing manner.
Stern-Ginossar N. & Ingolia N. T.
(2015)
Annual Review of Virology.
2,
p. 335-349
Viral genomes harbor a variety of unusual translational phenomena that allow them to pack coding information more densely and evade host restriction mechanisms imposed by the cellular translational apparatus. Annotating translated sequences within these genomes thus poses particular challenges, but identifying the full complement of proteins encoded by a virus is critical for understanding its life cycle and defining the epitopes it presents for immune surveillance. Ribosome profiling is an emerging technique for global analysis of translation that offers direct and experimental annotation of viral genomes. Ribosome profiling has been applied to two herpesvirus genomes, those of human cytomegalovirus and Kaposi's sarcoma-associated herpesvirus, revealing translated sequences within presumptive long non-coding RNAs and identifying other micropeptides. Synthesis of these proteins has been confirmed by mass spectrometry and by identifying T cell responses following infection. Ribosome profiling in other viruses will likely expand further our understanding of viral gene regulation and the proteome.
Stern-Ginossar N.
(2015)
Journal of Virology.
89,
12,
p. 6164-6166
Ribosome profiling is an emerging technique that uses deep sequencing to monitor translation in live cells. Studies using ribosome profiling have already provided novel insights into the identities and amounts of the proteins being produced in cells, as well as novel insights into the mechanism of protein synthesis and translation regulation. Application of ribosome profiling to cells infected with human cytomegalovirus and Kaposi's sarcoma-associated herpesvirus revealed unanticipated complexity in the coding capacity of herpesviruses. Here, I discuss these results and how the application of ribosome profiling to cells infected with other viruses can reveal novel insights into the process of infection.
Cohen Y. & Stern-Ginossar N.
(2014)
Seminars in Immunopathology.
36,
6,
p. 651-658
The herpesvirus human cytomegalovirus (HCMV) infects the majority of the worlds population, leading to severe diseases in millions of newborns and immunocompromised adults annually. During infection, HCMV extensively manipulates cellular gene expression to maintain conditions favorable for efficient viral propagation. Identifying the pathways that the virus relies on or subverts is of great interest as they have the potential to provide new therapeutic targets and to reveal novel principles in cell biology. Over the past years, high-throughput analyses have profoundly broadened our understanding of the processes that occur during HCMV infection. In this review, we will discuss these new findings and how they impact our understanding of the biology of HCMV.
Ingolia N. T., Brar G. A., Stern-Ginossar N., Harris M. S., Talhouarne G. J. S., Jackson S. E., Wills M. R. & Weissman J. S.
(2014)
Cell Reports.
8,
5,
p. 1365-1379
Ribosome profiling suggests that ribosomes occupy many regions of the transcriptome thought to be noncoding, including 5' UTRs and long noncoding RNAs (IncRNAs). Apparent ribosome footprints outside of protein-coding regions raise the possibility of artifacts unrelated to translation, particularly when they occupy multiple, overlapping open reading frames (ORFs). Here, we show hallmarks of translation in these footprints: copurification with the large ribosomal subunit, response to drugs targeting elongation, trinucleotide periodicity, and initiation at early AUGs. We develop a metric for distinguishing between 80S footprints and nonribosomal sources using footprint size distributions, which validates the vast majority of footprints outside of coding regions. We present evidence for polypeptide production beyond annotated genes, including the induction of immune responses following human cytomegalovirus (HCMV) infection. Translation is pervasive on cytosolic transcripts outside of conserved reading frames, and direct detection of this expanded universe of translated products enables efforts at understanding how cells manage and exploit its consequences.
Tsukerman P., Stern-Ginossar N., Yamin R., Ophir Y., Stanietsky A. M. N. & Mandelboim O.
(2014)
European Journal of Immunology.
44,
5,
p. 1517-1525
NK cells are innate immune lymphocytes that express a vast repertoire of germ-line encoded receptors for target recognition. These receptors include inhibitory and activating proteins, among the latter of which is CD16, a low affinity binding Fc receptor. Here, we show that human NK cells expand in response to stimulation with various tumor cell lines. We further demonstrate that the tumor-derived expansion of NK cells is accompanied by rapid, cell-dependent, changes in CD16 expression levels. We show that in NK cells expanded in response to the EBV-transformed cell line 721.221, CD16 is shed and therefore approximately half of the expanded 721.221-derived NK-cell population does not express CD16. We also show, in contrast, that in response to 1106mel cells, CD16 expression is maintained on the cell surface of the expanded NK cells due to an antibody-dependent mechanism. Our results may provide a basis for the selective expansion of NK cells that may be used for tumor immunotherapy.
Arias C., Weisburd B., Stern-Ginossar N., Mercier A., Madrid A. S., Bellare P., Holdorf M., Weissman J. S. & Ganem D.
(2014)
PLoS Pathogens.
10,
1,
e1003847.
Productive herpesvirus infection requires a profound, time-controlled remodeling of the viral transcriptome and proteome. To gain insights into the genomic architecture and gene expression control in Kaposi's sarcoma-associated herpesvirus (KSHV), we performed a systematic genome-wide survey of viral transcriptional and translational activity throughout the lytic cycle. Using mRNA-sequencing and ribosome profiling, we found that transcripts encoding lytic genes are promptly bound by ribosomes upon lytic reactivation, suggesting their regulation is mainly transcriptional. Our approach also uncovered new genomic features such as ribosome occupancy of viral non-coding RNAs, numerous upstream and small open reading frames (ORFs), and unusual strategies to expand the virus coding repertoire that include alternative splicing, dynamic viral mRNA editing, and the use of alternative translation initiation codons. Furthermore, we provide a refined and expanded annotation of transcription start sites, polyadenylation sites, splice junctions, and initiation/termination codons of known and new viral features in the KSHV genomic space which we have termed KSHV 2.0. Our results represent a comprehensive genome-scale image of gene regulation during lytic KSHV infection that substantially expands our understanding of the genomic architecture and coding capacity of the virus.
Gilbert L. A., Larson M. H., Morsut L., Liu Z., Brar G. A., Torres S. E., Stern-Ginossar N., Brandman O., Whitehead E. H., Doudna J. A., Lim W. A., Weissman J. S. & Qi L. S.
(2013)
Cell.
154,
2,
p. 442-451
The genetic interrogation and reprogramming of cells requires methods for robust and precise targeting of genes for expression or repression. The CRISPR-associated catalytically inactive dCas9 protein offers a general platform for RNA-guided DNA targeting. Here, we show that fusion of dCas9 to effector domains with distinct regulatory functions enables stable and efficient transcriptional repression or activation in human and yeast cells, with the site of delivery determined solely by a coexpressed short guide (sg)RNA. Coupling of dCas9 to a transcriptional repressor domain can robustly silence expression of multiple endogenous genes. RNA-seq analysis indicates that CRISPR interference (CRISPRi)-mediated transcriptional repression is highly specific. Our results establish that the CRISPR system can be used as a modular and flexible DNA-binding platform for the recruitment of proteins to a target DNA sequence, revealing the potential of CRISPRi as a general tool for the precise regulation of gene expression in eukaryotic cells.
Stern-Ginossar N., Weisburd B., Michalski A., Vu Thuy Khanh Le, Hein M. Y., Huang S., Ma M., Shen B., Qian S., Hengel H., Mann M., Ingolia N. T. & Weissman J. S.
(2012)
Science.
338,
6110,
p. 1088-1093
The human cytomegalovirus (HCMV) genome was sequenced 20 years ago. However, like those of other complex viruses, our understanding of its protein coding potential is far from complete. We used ribosome profiling and transcript analysis to experimentally define the HCMV translation products and follow their temporal expression. We identified hundreds of previously unidentified open reading frames and confirmed a fraction by means of mass spectrometry. We found that regulated use of alternative transcript start sites plays a broad role in enabling tight temporal control of HCMV protein expression and allowing multiple distinct polypeptides to be generated from a single genomic locus. Our results reveal an unanticipated complexity to the HCMV coding capacity and illustrate the role of regulated changes in transcript start sites in generating this complexity.
Tsukerman P., Stern-Ginossar N., Gur C., Glasner A., Nachmani D., Bauman Y., Yamin R., Vitenshtein A., Stanietsky N., Bar-Mag T., Lankry D. & Mandelboim O.
(2012)
Cancer Research.
72,
21,
p. 5463-5472
Natural killer cells (NK) are a component of innate immunity well known for their potent ability to kill virus-infected or neoplastically transformed cells following stimulation of the NK cell receptor NKG2D. One of the various ligands of NKG2D is MICB, a stress-induced ligand that has been found to be upregulated on the surface of tumor cells. However, there is little knowledge about how this upregulation may occur or how it may be selected against in tumors as a mechanism of immune escape. Here, we report that the metastasis-associated microRNA (metastamir) miR-10b directly binds to the 30 untranslated region of MICB and downregulates its expression. Notably, antagonizing miR-10b action enhanced NKG2D-mediated killing of tumor cells in vitro and enhanced clearance of tumors in vivo. Conversely, overexpression of miR-10b downregulated MICB and impaired elimination of tumor cells. Together, our results define MICB as a novel immune target of miR-10b, implying a direct link between metastasis capability and immune escape from NK cells. Cancer Res; 72(21); 5463-72. (C)2012 AACR.
Bauman Y., Nachmani D., Vitenshtein A., Tsukerman P., Drayman N., Stern-Ginossar N., Lankry D., Gruda R. & Mandelboim O.
(2011)
Cell Host and Microbe.
9,
2,
p. 93-102
The human polyoma viruses JCV and BKV establish asymptomatic persistent infection in 65%-90% of humans but can cause severe illness under immunosuppressive conditions. The mechanisms by which these viruses evade immune recognition are unknown. Here we show that a viral miRNA identical in sequence between JCV and BKV targets the stress-induced ligand ULBP3, which is a protein recognized by the killer receptor NKG2D. Consequently, viral miRNA-mediated ULBP3 downregulation results in reduced NKG2D-mediated killing of virus-infected cells by natural killer (NK) cells. Importantly, when the activity of the viral miRNA was inhibited during infection, NK cells killed the infected cells more efficiently. Because NKG2D is also expressed by various T cell subsets, we propose that JCV and BKV use an identical miRNA that targets ULBP3 to escape detection by both the innate and adaptive immune systems, explaining how these viruses remain latent without being eliminated by the immune system.
Stern-Ginossar N. & Mandelboim O.
(2010)
Natural Killer Cells
.
Thomson A. W. & Lotze M. T.(eds.).
p. 155-168
This chapter reviews the known NK receptors, their function, ligand specificity, and their overall contribution to NK cell recognition. Natural killer (NK) cells are bone marrow derived lymphocytes that are well-equipped for the destruction of tumor and virally infected cells without the need for prior antigen stimulation. Their killing machinery is quite complex and is determined by integrated signals obtained from activating and inhibitory receptors. Inhibitory receptors recognize molecules that are expressed on normal cells as a means of protecting healthy cells from attack by NK. This complex repertoire of NK cell receptors results in various NK cell subpopulations that can respond to bacteria, viruses, parasites, and tumor. Thus, although NK cells (unlike T and B cells) express only a limited number of receptors, the various combinations of receptors enable a remarkable diversity in NK cell activity. The prototypic inhibitory receptors recognize MHC class I molecules, but recent work has identified inhibitory receptors that have non-MHC-molecule ligands. The NK-activating receptors recognize pathogen-derived, stress-induced molecules and a series of still uncharacterized cellular ligands. Some of the future challenges in the NK field might include population dynamics, elucidating the precise threshold needed for NK cell activation and inhibition, and identifying the entire spectrum of NK cell inhibitory and activating receptors followed by the identification of their various physiological ligands.
Gur C., Porgador A., Elboim M., Gazit R., Mizrahi S., Stern-Ginossar N., Achdout H., Ghadially H., Dor Y., Nir T., Doviner V., Hershkovitz O., Mendelson M., Naparstek Y. & Mandelboim O.
(2010)
Nature Immunology.
11,
2,
p. 121-128
The mechanism of action of natural killer (NK) cells in type 1 diabetes is still unknown. Here we show that the activating receptor NKp46 recognizes mouse and human ligands on pancreatic beta cells. NK cells appeared in the pancreas when insulitis progressed to type 1 diabetes, and NKp46 engagement by beta cells led to degranulation of NK cells. NKp46-deficient mice had less development of type 1 diabetes induced by injection of a low dose of streptozotocin. Injection of soluble NKp46 proteins into nonobese diabetic mice during the early phase of insulitis and the prediabetic stage prevented the development of type 1 diabetes. Our findings demonstrate that NKp46 is essential for the development of type 1 diabetes and highlight potential new therapeutic modalities for this disease.
Manaster I., Gazit R., Goldman-Wohl D., Stern-Ginossar N., Mizrahi S., Yagel S. & Mandelboim O.
(2010)
Journal of Reproductive Immunology.
84,
1,
p. 1-7
NK cells specialize in killing tumor cells and vitally infected cells and also possess non-cytotoxic functions, which include secretion of a variety of cytokines and growth factors. Their activity is mediated by a vast repertoire of inhibitory and activating NK receptors. Recently, it was demonstrated that ligation of the Notch receptor plays a significant role not only in T cell development but also in human T cell and mouse NK cell activation. However, the involvement of Notch triggering in human NK cell activity has not yet been determined. Here we show that Notch1 and Notch2, but not Notch3 and Notch4, are expressed in human peripheral blood NK cells and in decidual NK cells. We demonstrate that in peripheral blood NK cells only the Notch ligand Delta4 could interact with Notch, whereas in decidual NK cells both Delta1 and Delta4 can interact with Notch. Finally, we show that Notch activation in these cells leads to increased secretion of IFN gamma. We therefore present here a new function of the Notch receptors as enhancers of peripheral blood NK cell and decidual NK cell functions. (C) 2009 Elsevier Ireland Ltd. All rights reserved.
Stanietsky N., Simic H., Arapovic J., Toporik A., Levy O., Novik A., Levine Z., Beiman M., Dassa L., Achdout H., Stern-Ginossar N., Tsukerman P., Jonjic S. & Mandelboim O.
(2009)
Proceedings Of The National Academy Of Sciences Of The United States Of America-Biological Sciences.
106,
42,
p. 17858-17863
NK cell cytotoxicity is controlled by numerous NK inhibitory and activating receptors. Most of the inhibitory receptors bind MHC class I proteins and are expressed in a variegated fashion. It was recently shown that TIGIT, a new protein expressed by T and NK cells binds to PVR and PVR-like receptors and inhibits T cell activity indirectly through the manipulation of DC activity. Here, we show that TIGIT is expressed by all human NK cells, that it binds PVR and PVRL2 but not PVRL3 and that it inhibits NK cytotoxicity directly through its ITIM. Finally, we show that TIGIT counter inhibits the NK-mediated killing of tumor cells and protects normal cells from NK-mediated cytoxicity thus providing an "alternative self'' mechanism for MHC class I inhibition.
Stern-Ginossar N., Saleh N., Goldberg M. D., Prichard M., Wolf D. G. & Mandelboim O.
(2009)
Journal of Virology.
83,
20,
p. 10684-10693
MicroRNAs (miRNAs) are expressed in a wide variety of organisms, ranging from plants to animals, and are key posttranscriptional regulators of gene expression. Virally encoded miRNAs are unique in that they could potentially target both viral and host genes. Indeed, we have previously demonstrated that a human cytomegalovirus (HCMV)-encoded miRNA, miR-UL112, downregulates the expression of a host immune gene, MICB. Remarkably, it was shown that the same miRNA also downregulates immediate-early viral genes and that its ectopic expression resulted in reduced viral replication and viral titers. The targets for most of the viral miRNAs, and hence their functions, are still unknown. Here we demonstrate that miR-UL112 also targets the UL114 gene, and we present evidence that the reduction of UL114 by miR-UL112 reduces its activity as uracil DNA glycosylase but only minimally affects virus growth. In addition, we show that two additional HCMV-encoded miRNAs, miR-US25-1 and miR-US25-2, reduce the viral replication and DNA synthesis not only of HCMV but also of other viruses, suggesting that these two miRNAs target cellular genes that are essential for virus growth. Thus, we suggest that in addition to miR-UL112, two additional HCMV miRNAs control the life cycle of the virus.
Stern-Ginossar N. & Mandelboim O.
(2009)
Immunology.
128,
1,
p. 1-6
NKG2D is one of the best characterized activating receptors and is expressed on natural killer cells and on various T-cell subsets. This receptor recognizes several different ligands that are induced by cellular stresses. In this review, we described the mechanisms controlling the expression of NKG2D ligands, with the emphasis on post-transcriptional and post-translational regulation.
Nachmani D., Stern-Ginossar N., Sarid R. & Mandelboim O.
(2009)
Cell Host and Microbe.
5,
4,
p. 376-385
Herpesviruses are known for their persistent lifelong latent infection, which is made possible by their vast repertoire of immune-evasion strategies. We have previously shown that a human cytomegalovirus (HCMV) microRNA represses expression of the stress-induced Natural Killer (NK) cell ligand, MICB, to escape recognition and consequent elimination by INK cells. Here, we show functional conservation among diverse microRNAs derived from different herpesviruses, including HCMV, Kaposi's sarcoma-associated herpesvirus (KSHV), and Epstein-Barr virus (EBV), in their ability to directly target MICB mRNA and reduce its expression. Although the various viral microRNAs share no sequence homology, they are functionally similar and target MICB at different yet adjacent sites during authentic viral infection. The finding that different herpesvirus microRNAs target MICB indicates that MICB plays a pivotal role in the clash between herpesviruses and humans.
Stern-Ginossar N., Gur C., Biton M., Horwitz E., Elboim M., Stanietsky N., Mandelboim M. & Mandelboim O.
(2008)
Nature Immunology.
9,
9,
p. 1065-1073
MICA and MICB are stress-induced ligands recognized by the activating receptor NKG2D. A microRNA encoded by human cytomegalovirus downregulates MICB expression by targeting a specific site in the MICB 3' untranslated region. As this site is conserved among different MICB alleles and a similar site exists in the MICA 3' untranslated region, we speculated that these sites are targeted by cellular microRNAs. Here we identified microRNAs that bound to these MICA and MICB 3' untranslated region sequences and obtained data suggesting that these microRNAs maintain expression of MICA and MICB protein under a certain threshold and facilitate acute upregulation of MICA and MICB during cellular stress. These microRNAs were overexpressed in various tumors and we demonstrate here that they aided tumor avoidance of immune recognition.
Manaster I., Mizrahi S., Goldman-Wohl D., Sela H. Y., Stern-Ginossar N., Lankry D., Gruda R., Hurwitz A., Bdolah Y., Haimov-Kochman R., Yagel S. & Mandelboim O.
(2008)
Journal of Immunology.
181,
3,
p. 1869-1876
NK cells populate the human endometrium before pregnancy. Unlike decidual NK cells that populate the decidua during pregnancy, the NK cells present in the human endometrium, before pregnancy, have not been fully characterized. In this study, we provide a detailed analysis of the origin, phenotype, and function of endometrial NK cells (eNK). We show that eNK cells have a unique receptor repertoire. In particular, they are negative for NKp30 and chemokine receptor expression, which distinguishes them from any other NK subset described so far. We further show that eNK cells lack NK-specific functional phenotype and activity such as cytokine secretion and cytotoxicity, before IL-15 stimulation. Following such stimulation, endometrial NK cells acquire phenotype and function that are similar to those of decidual NK cells. We therefore suggest that eNK cells are inactive cells (before IL-15 activation and in relation to the known NK activity) that are present in the endometrium before conception, waiting for pregnancy.
Mizrahi S., Yefenof E., Gross M., Attal P., Ben Yaakov A., Goldman-Wohl D., Maly B., Stern N., Katz G., Gazit R., Sionov R. V., Mandelboim O. & Chaushu S.
(2007)
Journal of Leukocyte Biology.
82,
5,
p. 1095-1105
Adenoids are part of the MALT. In the present study, we analyzed cell surface markers and cytolytic activity of adenoidal NK (A-NK) cells and compared them with NK cells derived from blood of the same donors (B-NK). NK cells comprised 0.67% (0.4-1.2%) of the total lymphoid population isolated from adenoids. The majority (median=92%) of the A-NK cells was CD56(bright) CD16(-). A-NK cells were characterized by the increased expression of activation-induced receptors. NKp44 was detected on >60%, CD25 on >40%, and HLA-DR on >50% of freshly isolated A-NK cells. Functional assays indicated that the cytotoxic machinery of A-NK is intact, and sensitive target cells are killed via natural cytotoxicity receptors, such as NKG2D. Carcinoembryonic antigen-related cell adhesion molecule I (CEACAM1; CD66) expression was up-regulated in 23% (median) of the A-NK cells by IL-2 activation but unchanged in B-NK cells. CEACAM1 inhibited the A-NK killing of target cells. CXCR4 was expressed on more than 40% A-NK cells prior to activation. Its ligand, CXCL12, was found in endothelial cells of the capillaries within the adenoid and in cells of the epithelial lining. In addition, A-NK cells migrated in vitro toward a gradient of CXCL 12 in a dose-responsive manner, suggesting a role for this chemokine in A-NK cell recruitment and trafficking. We conclude that the A-NK cells are unique in that they display an activated-like phenotype and are different from their CD16(-)B-NK cell counterparts. This phenotype presumably reflects the chronic interaction of A-NK cells with antigens penetrating the body through the nasal route.
Stern-Ginossar N., Nedvetzki S., Markel G., Gazit R., Betser-Cohen G., Achdout H., Aker M., Blumberg R. S., Davis D. M., Appelmelk B. & Mandelboim O.
(2007)
Journal of Immunology.
179,
7,
p. 4424-4434
The inhibition of NK cell killing is mainly mediated via the interaction of NK inhibitory receptors with MHC class I proteins. In addition, we have previously demonstrated that NK cells are inhibited in a class I MHC-independent manner via homophilic carcinoembryonic Ag (CEA) cell adhesion molecules (CEACAM1)-CEACAM1 and heterophilic CEACAM1-CEA interactions. However, the cross-talk between immune effector cells and their target cells is not limited to cell interactions per se, but also involves a specific exchange of proteins. The reasons for these molecular exchanges and the functional outcome of this phenomenon are still mostly unknown. In this study, we show that NK cells rapidly and specifically acquire CEA molecules from target cells. We evaluated the role of cytotoxicity in the acquisition of CEA and demonstrated it to be mostly killing independent. We further demonstrate that CEA transfer requires a specific interaction with an unknown putative NK cell receptor and that carbohydrates are probably involved in CEA recognition and acquisition by NK cells. Functionally, the killing of bulk NK cultures was inhibited by CEA-expressing cells, suggesting that this putative receptor is an inhibitory receptor.
Gruda R., Achdout H., Stern-Ginossar N., Gazit R., Betser-Cohen G., Manaster I., Katz G., Gonen-Gross T., Tirosh B. & Mandelboim O.
(2007)
Journal of Immunology.
179,
6,
p. 3655-3661
The activity of NK cells is regulated by activating receptors that recognize mainly stress-induced ligands and by inhibitory receptors that recognize mostly MHC class I proteins on target cells. Comparing the cytoplasmic tail sequences of various MHC class I proteins revealed the presence of unique cysteine residues in some of the MHC class I molecules which are absent in others. To study the role of these unique cysteines, we performed site specific mutagenesis, generating MHC class I molecules lacking these cysteines, and demonstrated that their expression on the cell surface was impaired. Surprisingly, we demonstrated that these cysteines are crucial for the surface binding of the leukocyte Ig-like receptor 1 inhibitory receptor to the MHC class I proteins, but not for the binding of the KIR2DL1 inhibitory receptor. In addition, we demonstrated that the cysteine residues in the cytoplasmic tail of MHC class I proteins are crucial for their egress from the endoplasmic reticulum and for their palmitoylation, thus probably affecting their expression on the cell surface. Finally, we show that the cysteine residues are important for proper extracellular conformation. Thus, although the interaction between leukocyte Ig-like receptor 1 and MHC class I proteins is formed between two extracellular surfaces, the intracellular components of MHC class I proteins play a crucial role in this recognition.
Stern-Ginossar N., Elefant N., Zimmermann A., Wolf D. G., Saleh N., Biton M., Horwitz E., Prokocimer Z., Prichard M., Hahn G., Goldman-Wohl D., Greenfield C., Yagel S., Hengel H., Altuvia Y., Margalit H. & Mandelboim O.
(2007)
Science.
317,
5836,
p. 376-381
Virally encoded microRNAs (miRNAs) have recently been discovered in herpesviruses. However, their biological roles are mostly unknown. We developed an algorithm for the prediction of miRNA targets and applied it to human cytomegalovirus miRNAs, resulting in the identification of the major histocompatibility complex class I-related chain B (MICB) gene as a top candidate target of hcmv-miR-UL112. MICB is a stress-induced ligand of the natural killer (NK) cell activating receptor NKG2D and is critical for the NK cell killing of virus-infected cells and tumor cells. We show that hcmv-miR-UL112 specifically down-regulates MICB expression during viral infection, leading to decreased binding of NKG2D and reduced killing by NK cells. Our results reveal a miRNA-based immunoevasion mechanism that appears to be exploited by human cytomegalovirus.
Stern N., Markel G., Arnon T. I., Gruda R., Wong H., Gray-Owen S. D. & Mandelboim O.
(2005)
Journal of Immunology.
174,
11,
p. 6692-6701
The NK killing activity is regulated by activating and inhibitory NK receptors. All of the activating ligands identified so far are either viral or stress-induced proteins. The class I MHC proteins are the ligands for most of the inhibitory NK receptors. However, in the past few years, several receptors have been identified that are able to inhibit NK killing independently of class I MHC recognition. We have previously demonstrated the existence of a novel inhibitory mechanism of NK cell cytotoxicity mediated by the homophilic carcinoembryonic Ag (CEA)-related cell adhesion molecule 1 (CEACAM1) interactions. In this study, we demonstrate that CEACAM1 also interacts heterophilically with the CEA protein. Importantly, we show that these heterophilic interactions of CEA and CEACAM1 inhibit the killing by NK cells. Because CEA is expressed on a wide range of carcinomas and commonly used as tumor marker, these results represent a novel role for the CEA protein enabling the escape of tumor cells from NK-mediated killing. We further characterize, for the first time, the CEACAM1-CEA interactions. Using functional and binding assays, we demonstrate that the N domains of CEACAM1 and CEA are crucial but not sufficient for both the CEACAM1-CEACAM1 homophilic and CEACAM1-CEA heterophilic interactions. Finally, we suggest that the involvement of additional domains beside the N domain in the heterophilic and homophilic interactions is important for regulating the balance between cis and trans interactions.