Reuven N., Winograd-Katz S., Barnea-Zohar M., Pri-Or A., Levin Y., Vacher J., Geiger B. & Elson A.
(2026)
Journal of Bone and Mineral Research.
zjaf196.
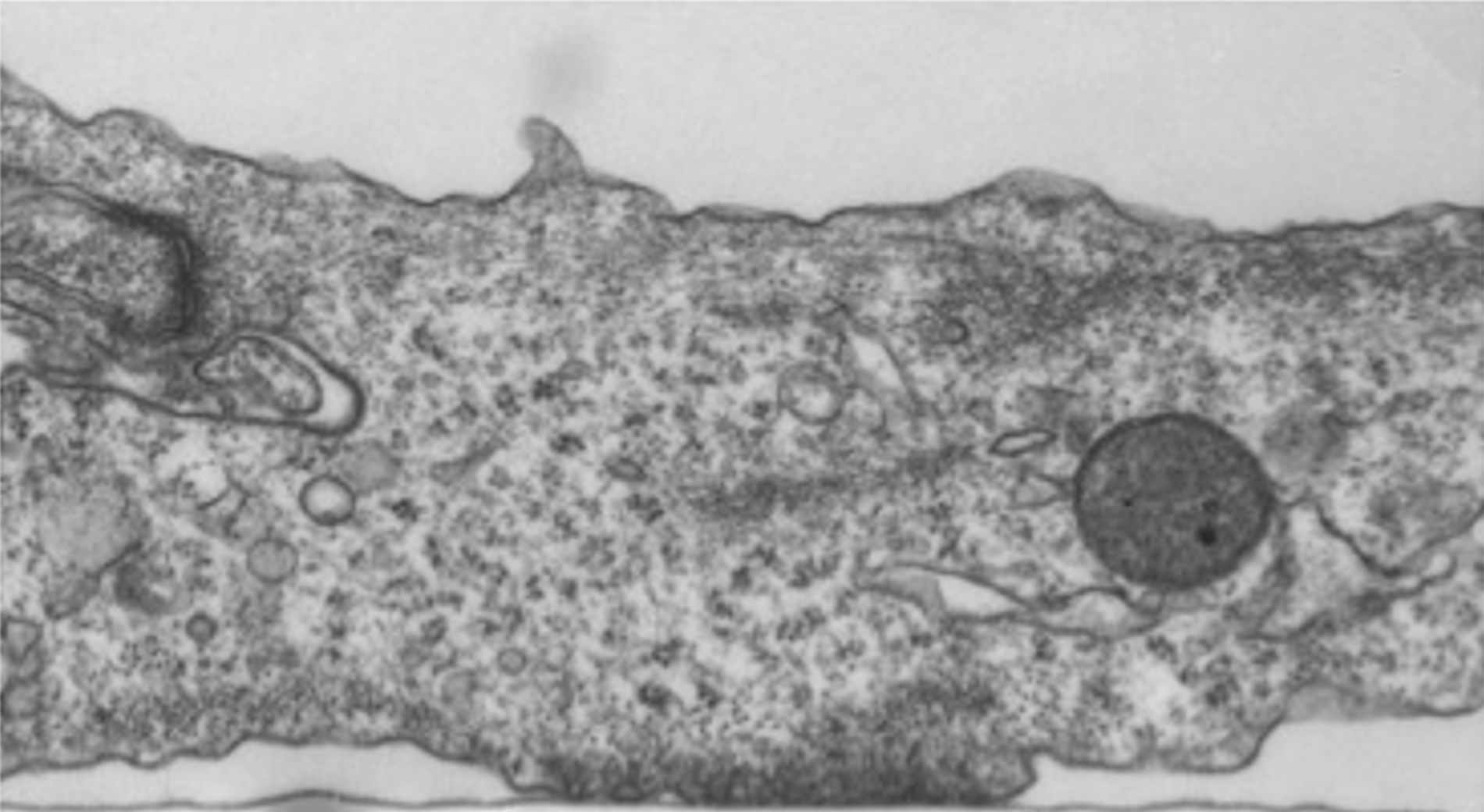
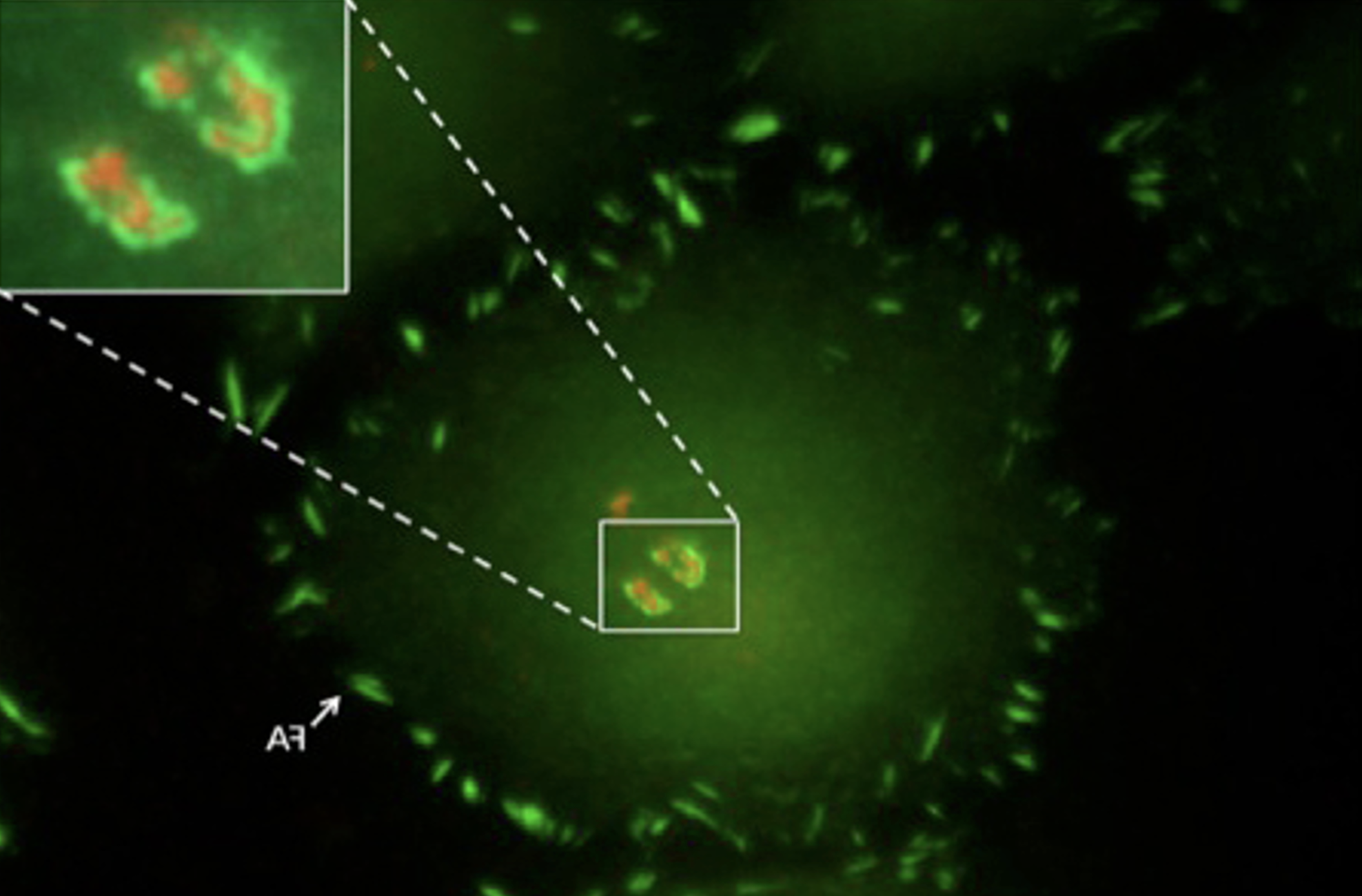
Bone-resorbing osteoclasts (OCLs) are large, multi-nucleated cells that are formed through well-regulated differentiation and cell fusion of monocyte-macrophage precursors. Abnormally increased or decreased OCL-mediated bone resorption perturbs bone structure and homeostasis and may lead to severe illnesses, such as osteoporosis and autosomal recessive osteopetrosis (ARO), respectively. Mutations in the intracellular trafficking-associated protein sorting nexin 10 (SNX10) lead to "OCL-rich" ARO, in which OCLs are inactive. Mature, SNX10-deficient murine OCLs fuse continuously to generate gigantic cells, in vitro and in vivo, unlike WT OCLs that stop fusing with each other upon maturation, indicating that SNX10 is required for both the resorptive activity of OCLs and the arrest of cell fusion upon maturation. Mutations in CLC-7 and OSTM1, which comprise the lysosomal voltage-gated Cl-/H+ exchanger, also induce OCL-rich ARO in humans and in mouse models, and are associated with the presence of large OCLs. In this study, we explored the molecular interplay between SNX10, CLC-7, and OSTM1 by comparing the phenotypes of cultured mouse OCLs lacking one of these proteins. We show that loss of each protein leads to the formation of similarly-gigantic OCLs in culture, due to deregulated fusion between mature OCLs that proceeds with similar kinetics. All 3 proteins co-localize in LAMP1-positive lysosomes, located at both perinuclear and peripheral regions of mature WT OCLs. SNX10-KO OCLs exhibit few peripheral lysosomes containing CLC-7 and OSTM1, indicating that SNX10 is required for regulating their trafficking to the cell periphery. CLC-7 and SNX10 physically interact with each other and loss of CLC-7 depletes peripheral OSTM1-containing lysosomes, indicating that CLC-7 is also required for this transport. Taken together, these findings indicate that SNX10 and CLC-7 regulate the subcellular distribution of lysosomes containing CLC-7 and OSTM1, thereby establishing a functional link between these 3 proteins that controls both the fusion and functionality of mature OCLs.Loss of the proteins SNX10, CLC-7, or OSTM1 in autosomal recessive osteopetrosis abolishes bone degradation by osteoclasts (OCLs), leading to morbidly-increased bone mass. The molecular interplay between these proteins is unknown. We show that OCLs lacking SNX10, CLC-7, or OSTM1 fuse continuously to form gigantic, inactive cells. The 3 proteins co-localize in subcellular organelles called lysosomes, SNX10 and CLC-7 physically associate, and both are essential for positioning lysosomes at the cell periphery, where they secrete bone-degrading compounds. Disrupting lysosomal activity or peripheral positioning by loss of SNX10, CLC-7, or OSTM1 blocks OCL-mediated bone resorption activity and deregulates OCL fusion.
Perera W. R., Ansari A., Nowell C., Geiger B., Voelcker N. H., Frith J. E. & Cadarso V. J.
(2025)
Small.
22,
7,
e08899.
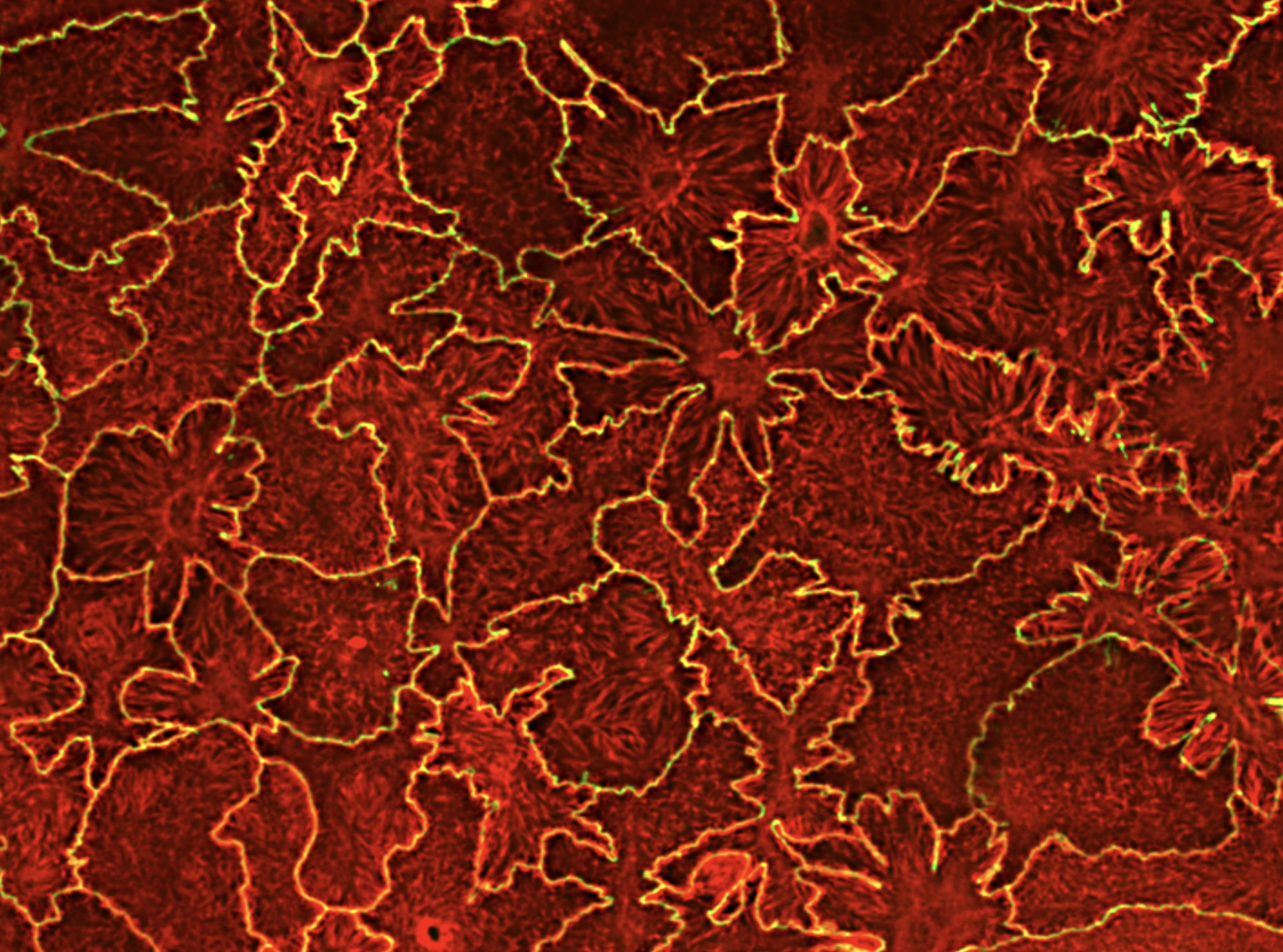
Understanding cellular response to mechanical cues in three-dimensional (3D) environments remains a central challenge in cell biology. Shape and force distribution are key regulators of mechanosensing. In vivo, cells are embedded in 3D environments where force transmission and cytoskeletal behavior differ markedly from two-dimensional systems. However, current tools lack the resolution to precisely control single-cell geometry or quantify traction forces in defined 3D contexts. Here, a direct laser writing-based platform is presented that fabricates microscale cage structures capable of confining individual mesenchymal stem cells in defined 3D geometries while enabling high-resolution traction force measurements. The system allows independent control of cell volume and shape, and captures nanowire deflection as a readout of cell-generated forces at varying heights. Using this platform, this study reveals that 3D shape alone, modulates cytoskeletal organization, contractility, and localization of the mechanosensitive transcription factor Yes-associated protein (YAP) in a shape and time dependent manner. Square cages induced previously unreported vertical actin fibers and corner-enriched myosin accumulation, suggesting that pointed 3D geometry alters internal force distribution. Delayed YAP nuclear translocation indicates a time-sensitive mechanotransduction response to 3D confinement. Altogether, this platform offers a tunable 3D confinement tool and new insights into how shape alone direct cellular force architecture and mechanosensitive signaling.
Chung W., Boujemaa-Paterski R., Winograd-Katz S., Eibauer M., Geiger B. & Medalia O.
(2025)
BioRxiv.
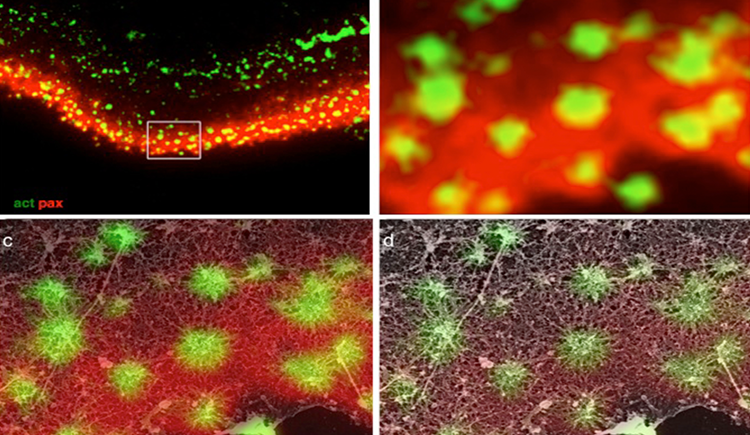
Focal adhesions (FAs) are dynamic macromolecular assemblies that anchor the actin cytoskeleton to the extracellular matrix via integrin receptors, thereby regulating cell morphology and migration. Although FA maturation and organization have been extensively studied, it remains unclear how regulatory proteins influence the 3D architecture of FAs. Here, we show that loss of the vasodilator-stimulated phosphoprotein (VASP) impairs adhesion dynamics. We employed CRISPR/Cas9-mediated knockout of VASP and/or the mechanosensitive adaptor protein zyxin to investigate their respective roles in actintextendashadhesion coupling. Loss of VASP and zyxin correlates with altered FA morphology and impaired dynamics. Using cryo-electron tomography (cryo-ET), we resolved the polarity of individual actin filaments associated with FAs and identified a contractility-related actin layer enriched with tropomyosin. VASP and zyxin are required for the assembly of dense and aligned actin bundles with uniform polarity, oriented with their barbed ends towards the cell edge. In contrast, the tropomyosin-decorated dorsal actin layer remains unaffected by these perturbations. Our findings reveal distinct, layered architectures within FAs and underscore the cooperative role of VASP and zyxin in stabilizing the organization of actin filaments at functional adhesion sites.Competing Interest StatementThe authors have declared no competing interest.