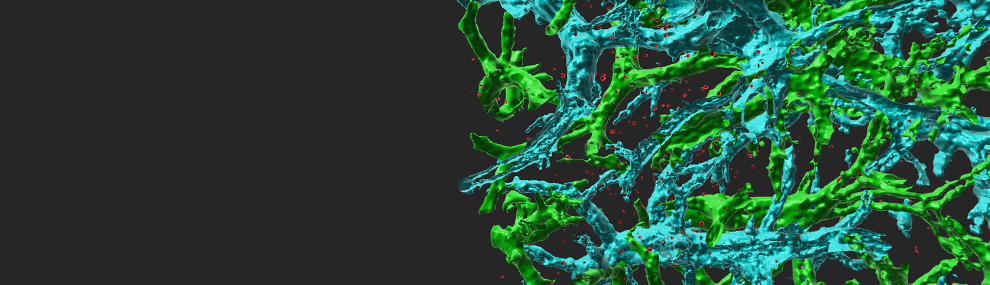
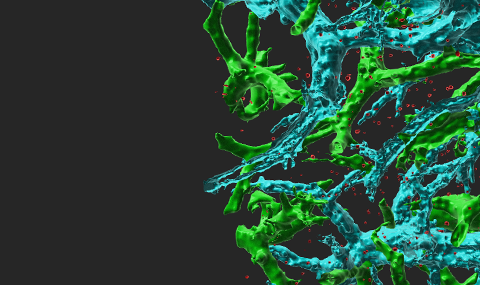
Endothelial chemokines are instrumental for integrin mediated lymphocyte adhesion and TEM through vascular barriers. Addressing how chemokine signals promote these two processes using flow chamber assays, we found that following arrest, both T and B lymphocytes rapidly crawl on inflamed endothelium in an LFA-1 and chemokine dependent manner. We also found that endothelial presented chemokines trigger lymphocyte crawling by stimulating high affinity (HA) LFA-1 at numerous focal submicron dots underneath the entire adhesive zone reversibly generated by the rapidly crawling lymphocyte. Surface bound chemokines and ICAM-1 are both necessary and sufficient for these quantal LFA-1 dots to form and rapidly dissociate under physiological shear flow. Interestingly, chemokine stimulated VLA-4 remains clustered at the rear of crawling lymphocytes, shortly after microvillar collapse and in proximity to other microvillar surface receptors like PSGL-1 and CD43. Notably, shear forces applied on crawling lymphocytes increased the density of both LFA-1 dots and of adhesive filopodia extended from the base of the crawling lymphocyte. Strikingly, a fraction of these filopodia became highly invasive into the endothelial cell body even prior to transendothelial crossing. These filopodia are not classical podosomes recently observed in T blasts crossing activated endothelium. The density of invasive filopodia increased with the magnitude of chemokine and ICAM-1 signals and closely correlated with the ability of T cells to successfully transmigrate upon reaching paracellular junctions. During TEM, T cells extend large subluminal lamellopodia, enriched with HA-LFA-1 and subluminal ICAM-1, suggesting that basal endothelial ICAM-1 guides T cell TEM. Similar LFA-1 dots have not yet been observed in vivo due to poor resolution of integrin imaging, but several EM reports detected invasive protrusions underneath lymphocytes associated with multiple endothelial beds in vivo. We propose that HA-LFA-1 dots are key shear resistant adhesive units which give rise to adhesive and invasive filopodia that scan the endothelial surface for apical and junctional guidance signals for diapedesis. Our recent results also suggest the involvement of the CDC42 GTPase in this chemokine triggered, shear force-facilitated LFA-1-dependent process. In addition, we were among the first groups to identify novel transcellular routes taken by neutrophils and small subsets of T effector cells to cross endothelial barriers (Movie 1).
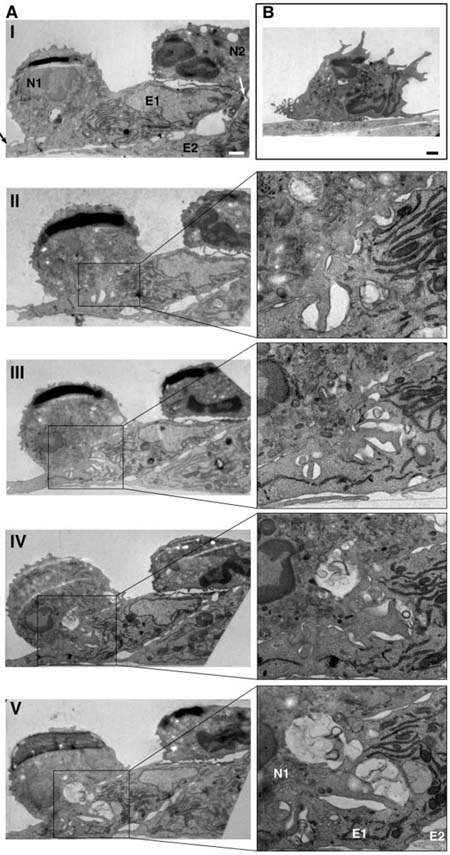
Figure 1 Shear stress induces massive apical invaginations in neutrophils and lymphocytes Left: TEM through PAF-presenting, TNAFα-activated HUVEC. A. Ultrastructural analysis of neutrophils migrating under shear flow across PAF-presenting HUVEC prestimulated with TNFα. Shown are serial cross sections of two neutrophils adhering to and migrating across neighboring ECs. B. A representative neutrophil left under shear on IL-8 presenting HUVEC prestimulated with TNFα. Right: Two T lymphocytes sending invasive filopodia into the body of a cytokine stimulated endothelial cell in the presence of shear forces. The density of these filopodia is increased by up to 5 fold in the presence of maximal stimulatory conditions (shear stress, high levels of apical chemokines).
The Rho/Rac GTPases are in situ triggered by signals from endothelial chemokines signals, and function as key regulators of leukocyte integrin activation and motility. In lymphocytes, a major portion of chemokine-mediated Rac activation depends on the CDM adaptor DOCK2. Using a novel ex vivo model for real time analysis of murine leukocyte subsets, we previously identified a specialized role for this adaptor in chemokine-triggered integrin-independent lymphocyte motility on endothelial and extracellular matrix barriers but not in chemokine-triggered integrin-mediated adhesiveness or the transmigration of lymphocytes through a chemokine-presenting endothelial barrier. We have found that DOCK2 is also crucial for activation of a subset of Rac mediated activities which control the ability of lymphocytes to crawl away from the arrest site to endothelial junctions, a process mediated primarily by their LFA-1 integrin. Although implicated in integrin activation, Rap-1 is not required for this integrin-independent chemokine triggered lymphocyte motility.